Simple Alignment Example¤
This example demonstrates basic differentiable sequence alignment using DiffBio's Smith-Waterman operator.
Setup¤
import jax
import jax.numpy as jnp
from diffbio.operators.alignment import (
SmoothSmithWaterman,
SmithWatermanConfig,
create_dna_scoring_matrix,
)
Create the Aligner¤
# Create a DNA scoring matrix
# match=2.0 for identical nucleotides, mismatch=-1.0 for different ones
scoring_matrix = create_dna_scoring_matrix(match=2.0, mismatch=-1.0)
# Configure the Smith-Waterman aligner
config = SmithWatermanConfig(
temperature=1.0, # Smoothness parameter (1.0 is balanced)
gap_open=-10.0, # Penalty for starting a gap
gap_extend=-1.0, # Penalty per additional gap position
)
# Create the aligner
aligner = SmoothSmithWaterman(config, scoring_matrix=scoring_matrix)
Encode Sequences¤
DiffBio uses one-hot encoding for sequences. The DNA alphabet is A=0, C=1, G=2, T=3.
def one_hot_dna(sequence_string):
"""Convert DNA sequence string to one-hot encoding."""
mapping = {'A': 0, 'C': 1, 'G': 2, 'T': 3}
indices = jnp.array([mapping[c] for c in sequence_string])
return jnp.eye(4)[indices]
# Example sequences
seq1 = one_hot_dna("ACGTACGT") # 8 nucleotides
seq2 = one_hot_dna("ACATACGT") # Different at position 3 (G->A)
print(f"Sequence 1 shape: {seq1.shape}") # (8, 4)
print(f"Sequence 2 shape: {seq2.shape}") # (8, 4)
Output:
Perform Alignment¤
# Run the alignment via apply()
data = {"seq1": seq1, "seq2": seq2}
result, _, _ = aligner.apply(data, {}, None)
print(f"Alignment score: {result['score']:.4f}")
print(f"Alignment matrix shape: {result['alignment_matrix'].shape}") # (9, 9)
print(f"Soft alignment shape: {result['soft_alignment'].shape}") # (8, 8)
Output:
Interpret Results¤
Alignment Score¤
The alignment score indicates overall similarity:
Output:
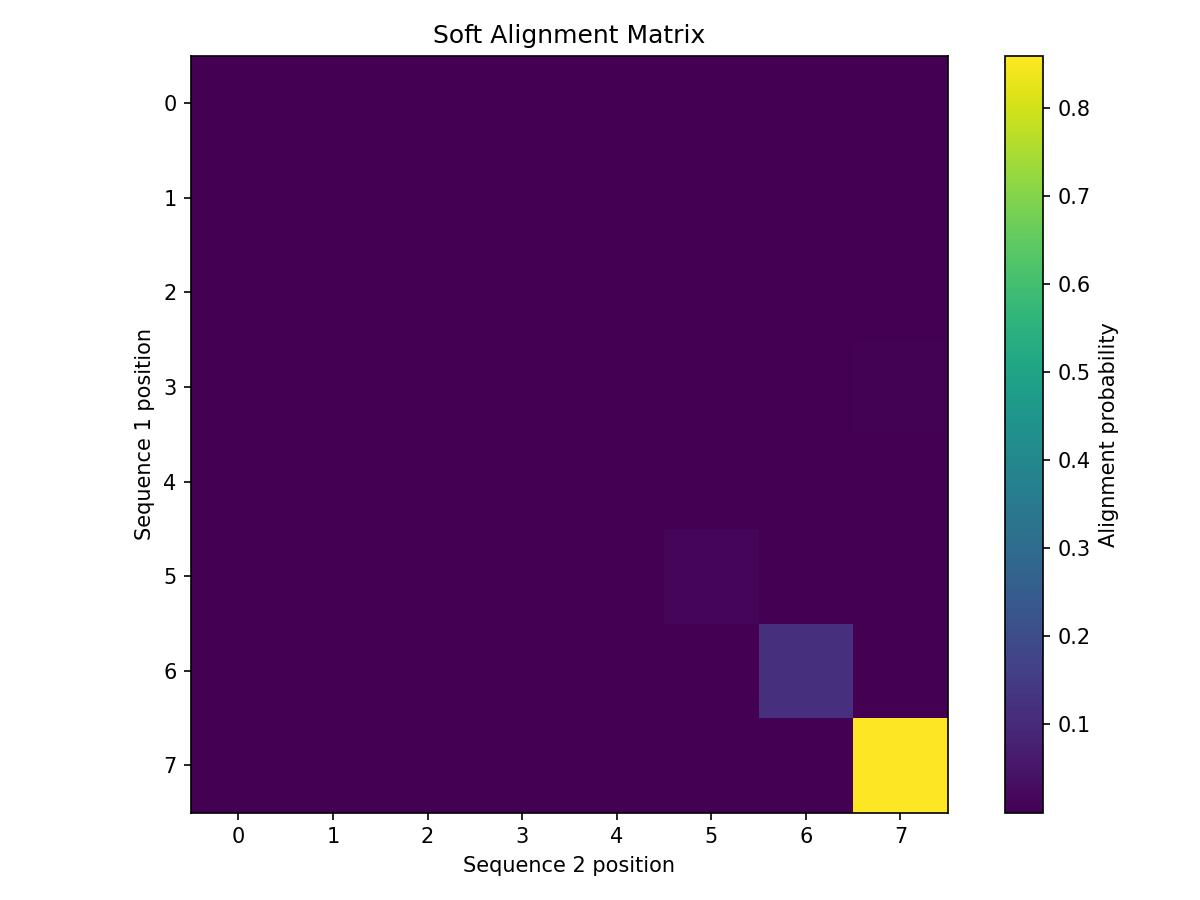
Soft Alignment Matrix¤
The soft alignment shows position correspondences as probabilities:
# Find most likely corresponding positions
best_matches = jnp.argmax(result["soft_alignment"], axis=1)
print(f"Position correspondences: {best_matches}")
# Position i in seq1 corresponds to position best_matches[i] in seq2
Output:
Visualization (Optional)¤
import matplotlib.pyplot as plt
plt.figure(figsize=(8, 6))
plt.imshow(result["soft_alignment"], cmap='viridis')
plt.colorbar(label='Alignment probability')
plt.xlabel('Sequence 2 position')
plt.ylabel('Sequence 1 position')
plt.title('Soft Alignment Matrix')
plt.show()
Compute Gradients¤
The key feature of DiffBio is differentiability:
# Define a loss function based on alignment score
def alignment_loss(scoring_matrix, seq1, seq2):
config = SmithWatermanConfig(temperature=1.0)
aligner = SmoothSmithWaterman(config, scoring_matrix=scoring_matrix)
data = {"seq1": seq1, "seq2": seq2}
result, _, _ = aligner.apply(data, {}, None)
return -result["score"] # Negative because we want to maximize
# Compute gradient w.r.t. scoring matrix
grad_fn = jax.grad(alignment_loss)
grads = grad_fn(scoring_matrix, seq1, seq2)
print("Scoring matrix gradients:")
print(grads)
Output:
Scoring matrix gradients:
[[-1.8247273e+00 -1.9172340e-07 -2.9888235e-15 -7.5641262e-09]
[-5.5532780e-07 -1.9372165e+00 -1.4243892e-10 -5.4443811e-12]
[-9.5245820e-01 -3.6374666e-07 -9.9998236e-01 -5.4135889e-08]
[-6.1676126e-08 -1.7161713e-12 -2.0101693e-11 -1.9935606e+00]]
The gradients tell us how to adjust the scoring matrix to improve alignment.
Using the Datarax Interface¤
For batch processing, use the apply() method:
# Prepare data as dictionary
data = {
"seq1": seq1,
"seq2": seq2,
}
# Apply operator
result_data, state, metadata = aligner.apply(data, {}, None)
# Access results
print(f"Score: {result_data['score']:.4f}")
print(f"Keys: {result_data.keys()}")
Output:
Batch Processing with vmap¤
Process multiple sequence pairs in parallel:
# Create batch of sequence pairs
batch_seq1 = jnp.stack([
one_hot_dna("ACGTACGT"),
one_hot_dna("TTTTAAAA"),
one_hot_dna("GCGCGCGC"),
])
batch_seq2 = jnp.stack([
one_hot_dna("ACATACGT"),
one_hot_dna("TTTTAAAG"),
one_hot_dna("GCGCATGC"),
])
# Vectorize alignment
def align_pair(s1, s2):
data = {"seq1": s1, "seq2": s2}
result, _, _ = aligner.apply(data, {}, None)
return result["score"]
batch_align = jax.vmap(align_pair)
batch_scores = batch_align(batch_seq1, batch_seq2)
print(f"Batch scores: {batch_scores}")
Output:
Temperature Effects¤
The temperature parameter controls the smoothness of the alignment:
temperatures = [0.1, 1.0, 5.0, 10.0]
for temp in temperatures:
config = SmithWatermanConfig(temperature=temp)
aligner = SmoothSmithWaterman(config, scoring_matrix=scoring_matrix)
data = {"seq1": seq1, "seq2": seq2}
result, _, _ = aligner.apply(data, {}, None)
print(f"Temperature {temp}: score = {result['score']:.4f}")
Output:
Temperature 0.1: score = 13.0000
Temperature 1.0: score = 13.1916
Temperature 5.0: score = 22.2542
Temperature 10.0: score = 58.6904
- Low temperature (0.1): Near-discrete, sharp alignment
- High temperature (10.0): Very smooth, gradients flow more easily
Learning Optimal Parameters¤
Train the scoring matrix for a specific task:
import optax
from flax import nnx
# Initial scoring matrix
initial_scoring = create_dna_scoring_matrix(match=2.0, mismatch=-1.0)
# Create aligner with learnable scoring
config = SmithWatermanConfig(temperature=1.0)
aligner = SmoothSmithWaterman(config, scoring_matrix=initial_scoring)
# Define target (we want seq1 and seq2 to align with score > 10)
target_score = 10.0
def loss(aligner, seq1, seq2, target):
data = {"seq1": seq1, "seq2": seq2}
result, _, _ = aligner.apply(data, {}, None)
return (result["score"] - target) ** 2
# Optimizer
params = nnx.state(aligner, nnx.Param)
optimizer = optax.adam(learning_rate=0.1)
opt_state = optimizer.init(params)
# Training loop
for step in range(100):
loss_val, grads = jax.value_and_grad(loss)(aligner, seq1, seq2, target_score)
params = nnx.state(aligner, nnx.Param)
updates, opt_state = optimizer.update(grads, opt_state, params)
nnx.update(aligner, optax.apply_updates(params, updates))
if step % 20 == 0:
print(f"Step {step}: loss = {loss_val:.4f}")
# Final score
final_data = {"seq1": seq1, "seq2": seq2}
final_result, _, _ = aligner.apply(final_data, {}, None)
print(f"Final score: {final_result['score']:.4f} (target: {target_score})")
Output:
Step 0: loss = 10.1866
Step 20: loss = 0.6327
Step 40: loss = 0.0532
Step 60: loss = 0.0085
Step 80: loss = 0.0015
Final score: 10.0036 (target: 10.0)
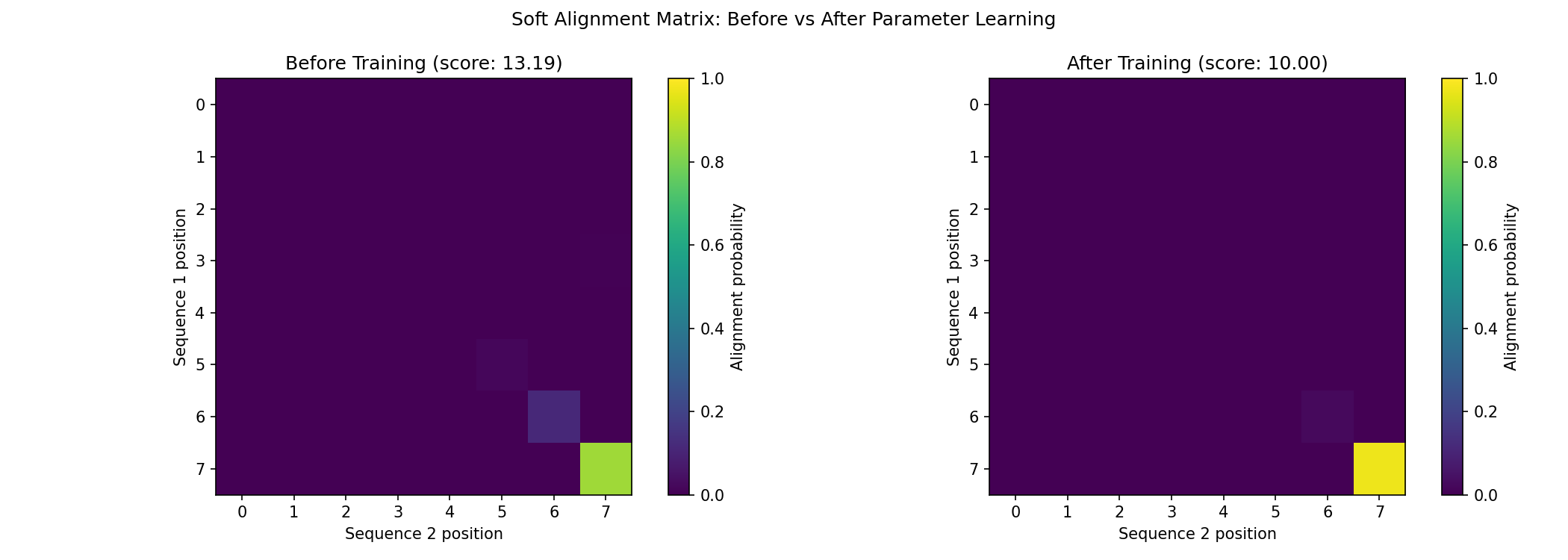
Visualize Learned Alignment¤
Compare the soft alignment before and after training:
# Get alignment after training
final_data = {"seq1": seq1, "seq2": seq2}
final_result, _, _ = aligner.apply(final_data, {}, None)
# Visualization comparing before and after
fig, axes = plt.subplots(1, 2, figsize=(14, 5))
# Before training (using initial aligner)
initial_aligner = SmoothSmithWaterman(config, scoring_matrix=initial_scoring)
initial_result, _, _ = initial_aligner.apply(final_data, {}, None)
im1 = axes[0].imshow(initial_result["soft_alignment"], cmap='viridis', vmin=0, vmax=1)
axes[0].set_xlabel('Sequence 2 position')
axes[0].set_ylabel('Sequence 1 position')
axes[0].set_title(f'Before Training (score: {initial_result["score"]:.2f})')
plt.colorbar(im1, ax=axes[0], label='Alignment probability')
# After training
im2 = axes[1].imshow(final_result["soft_alignment"], cmap='viridis', vmin=0, vmax=1)
axes[1].set_xlabel('Sequence 2 position')
axes[1].set_ylabel('Sequence 1 position')
axes[1].set_title(f'After Training (score: {final_result["score"]:.2f})')
plt.colorbar(im2, ax=axes[1], label='Alignment probability')
plt.suptitle('Soft Alignment Matrix: Before vs After Parameter Learning')
plt.tight_layout()
plt.show()
Summary¤
This example demonstrated:
- Creating a differentiable Smith-Waterman aligner
- One-hot encoding DNA sequences
- Performing smooth alignments
- Computing gradients for optimization
- Using vmap for batch processing
- Learning optimal alignment parameters
Next Steps¤
- Try Pileup Generation for variant calling
- See Variant Calling Pipeline for a complete workflow