MolNet Data Loading¤
This example demonstrates how to load MoleculeNet benchmark datasets using DiffBio's MolNetSource data source.
Overview¤
MoleculeNet provides standardized molecular property datasets for benchmarking machine learning models. DiffBio integrates these datasets through the MolNetSource class, which provides:
- Automatic downloading and caching
- Train/validation/test splits
- Task type information (classification/regression)
- Seamless integration with DiffBio operators
Prerequisites¤
Loading a Dataset¤
Load the BBBP (Blood-Brain Barrier Penetration) dataset:
# Configure the data source
config = MolNetSourceConfig(
dataset_name="bbbp",
split="train",
download=True,
)
# Create the source
source = MolNetSource(config)
# Check dataset properties
print(f"Dataset: {config.dataset_name}")
print(f"Number of molecules: {len(source)}")
print(f"Task type: {source.task_type}")
print(f"Number of tasks: {source.n_tasks}")
Output:
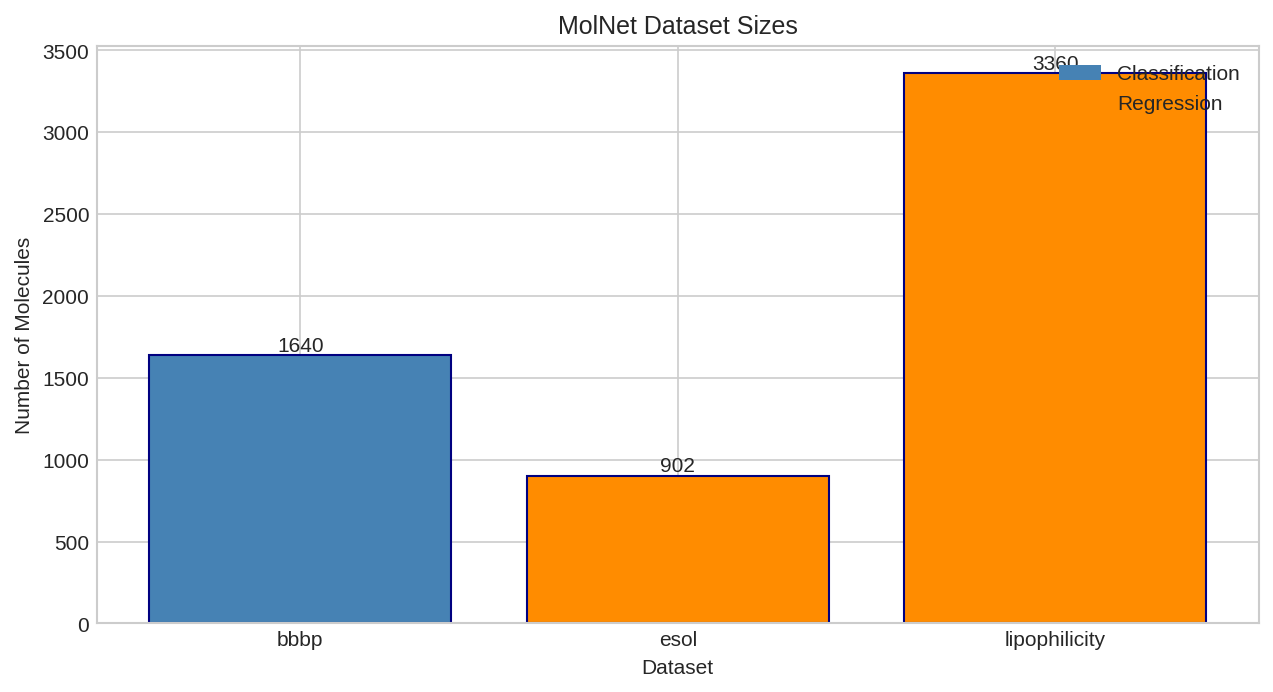
Size comparison of various MolNet benchmark datasets. Classification datasets (BBBP, HIV, Tox21) and regression datasets (ESOL, FreeSolv, Lipophilicity) vary widely in size.
Accessing Molecules¤
Iterate through molecules to access SMILES and labels:
# Show first few molecules
print("First 3 molecules:")
for i in range(3):
element = source[i]
smiles = element.data["smiles"]
label = element.data["y"]
print(f" Molecule {i}: {smiles[:40]}... | BBB+: {label}")
Output:
First 3 molecules:
Molecule 0: [Cl].CC(C)NCC(O)COc1cccc2ccccc12 | BBB+: 1.0
Molecule 1: C(=O)(OC(C)(C)C)CCCc1ccc(cc1)N(CCCl)CCCl | BBB+: 1.0
Molecule 2: c12c3c(N4CCN(C)CC4)c(F)cc1c(c(C(O)=O)cn2... | BBB+: 1.0
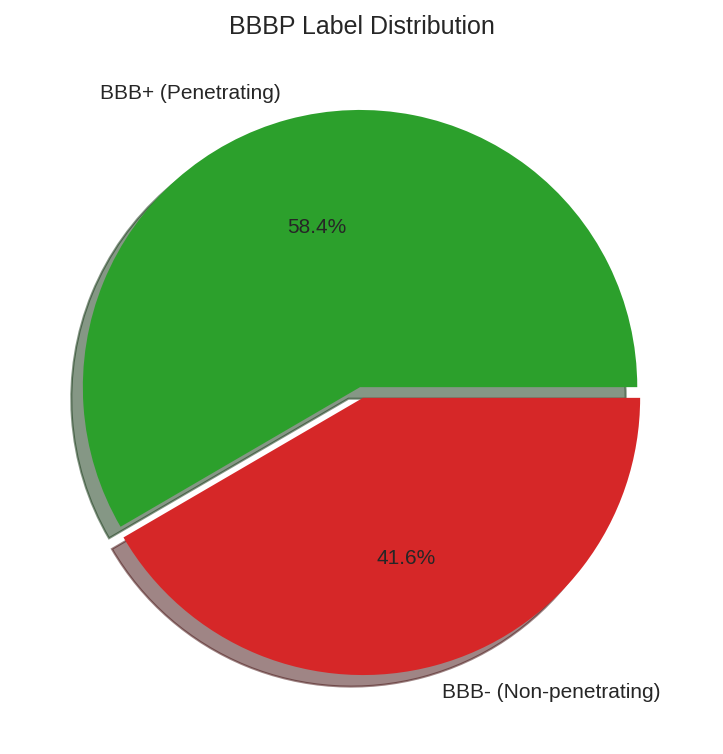
Label distribution for the BBBP dataset showing the class imbalance (more BBB+ than BBB- molecules).
Available Datasets¤
MolNetSource supports the following MoleculeNet datasets:
| Dataset | Task Type | Tasks | Description |
|---|---|---|---|
bbbp |
Classification | 1 | Blood-Brain Barrier Penetration |
bace |
Classification | 1 | BACE-1 binding affinity |
hiv |
Classification | 1 | HIV replication inhibition |
tox21 |
Classification | 12 | Toxicity assays |
toxcast |
Classification | 617 | EPA ToxCast assays |
sider |
Classification | 27 | Side effect database |
clintox |
Classification | 2 | Clinical toxicity |
esol |
Regression | 1 | Aqueous solubility |
freesolv |
Regression | 1 | Free solvation energy |
lipophilicity |
Regression | 1 | Octanol/water partition |
qm7 |
Regression | 1 | Electronic properties |
qm8 |
Regression | 16 | Quantum mechanics properties |
qm9 |
Regression | 12 | Quantum mechanics properties |
Element Structure¤
Each element from MolNetSource contains:
element = source[0]
# Data dictionary
element.data["smiles"] # SMILES string
element.data["y"] # Label(s) - scalar or array for multi-task
# Element metadata
element.state # Element state (empty dict by default)
element.metadata # Additional metadata
Using with Splitters¤
Combine with DiffBio splitters for proper train/test evaluation:
from diffbio.splitters import ScaffoldSplitter, ScaffoldSplitterConfig
# Create scaffold splitter for drug discovery
split_config = ScaffoldSplitterConfig(
train_frac=0.8,
valid_frac=0.1,
test_frac=0.1,
)
splitter = ScaffoldSplitter(split_config)
# Split the data
result = splitter.split(source)
print(f"Train: {len(result.train_indices)} molecules")
print(f"Valid: {len(result.valid_indices)} molecules")
print(f"Test: {len(result.test_indices)} molecules")
Comparison of random vs scaffold splitting showing train/valid/test distribution.
Next Steps¤
- Molecular Fingerprints - Generate fingerprints from molecules
- Scaffold Splitting - Proper data splitting for drug discovery
- Drug Discovery Workflow - Complete training workflow