Pileup Generation Example¤
This example demonstrates how to generate differentiable pileups from sequencing reads.
Setup¤
import jax
import jax.numpy as jnp
from diffbio.operators.variant import DifferentiablePileup, PileupConfig
Create the Pileup Operator¤
# Configure pileup generation
config = PileupConfig(
reference_length=100, # Length of reference sequence
use_quality_weights=True, # Weight by quality scores
)
# Create operator
pileup_op = DifferentiablePileup(config)
Prepare Read Data¤
Pileup generation requires three inputs:
- reads: One-hot encoded reads
(num_reads, read_length, 4) - positions: Starting position of each read
(num_reads,) - quality: Phred quality scores
(num_reads, read_length)
# Simulate some reads
num_reads = 50
read_length = 30
reference_length = 100
# Generate random one-hot reads
key = jax.random.PRNGKey(42)
k1, k2, k3 = jax.random.split(key, 3)
# Random nucleotide indices, then one-hot encode
read_indices = jax.random.randint(k1, (num_reads, read_length), 0, 4)
reads = jax.nn.one_hot(read_indices, 4)
print(f"Reads shape: {reads.shape}") # (50, 30, 4)
Output:
# Random starting positions (ensuring reads fit within reference)
positions = jax.random.randint(k2, (num_reads,), 0, reference_length - read_length)
print(f"Positions shape: {positions.shape}") # (50,)
print(f"Position range: {positions.min()} to {positions.max()}")
Output:
# Random quality scores (Phred scale, typically 0-40)
quality = jax.random.uniform(k3, (num_reads, read_length), minval=10.0, maxval=40.0)
print(f"Quality shape: {quality.shape}") # (50, 30)
print(f"Quality range: {quality.min():.1f} to {quality.max():.1f}")
Output:
Generate Pileup¤
Using compute_pileup directly¤
result = pileup_op.compute_pileup(reads, positions, quality, reference_length)
pileup = result["pileup"]
print(f"Pileup shape: {pileup.shape}") # (100, 4)
print(f"Pileup at position 50: {pileup[50]}")
print(f"Sum at position 50 (should be ~1): {pileup[50].sum():.4f}")
Output:
Pileup shape: (100, 4)
Pileup at position 50: [0.25692227 0.24718648 0.22821887 0.26767245]
Sum at position 50 (should be ~1): 1.0000
Using the Datarax interface¤
data = {
"reads": reads,
"positions": positions,
"quality": quality,
}
result, state, metadata = pileup_op.apply(data, {}, None)
pileup = result["pileup"]
print(f"Pileup shape: {pileup.shape}")
Output:
Interpret the Pileup¤
The pileup is a probability distribution over nucleotides at each position:
# Check nucleotide distribution at position 50
pos = 50
print(f"Position {pos}:")
print(f" A: {pileup[pos, 0]:.4f}")
print(f" C: {pileup[pos, 1]:.4f}")
print(f" G: {pileup[pos, 2]:.4f}")
print(f" T: {pileup[pos, 3]:.4f}")
Output:
Find Dominant Nucleotide¤
# Most likely nucleotide at each position
dominant = jnp.argmax(pileup, axis=-1)
nucleotides = ['A', 'C', 'G', 'T']
consensus = ''.join([nucleotides[i] for i in dominant])
print(f"Consensus sequence: {consensus[:50]}...")
Output:
Visualize Coverage¤
# Compute coverage (number of reads overlapping each position)
def compute_coverage(positions, read_length, reference_length):
coverage = jnp.zeros(reference_length)
for i in range(len(positions)):
pos = positions[i]
coverage = coverage.at[pos:pos+read_length].add(1)
return coverage
coverage = compute_coverage(positions, read_length, reference_length)
print(f"Coverage range: {coverage.min():.0f} to {coverage.max():.0f}")
Output:
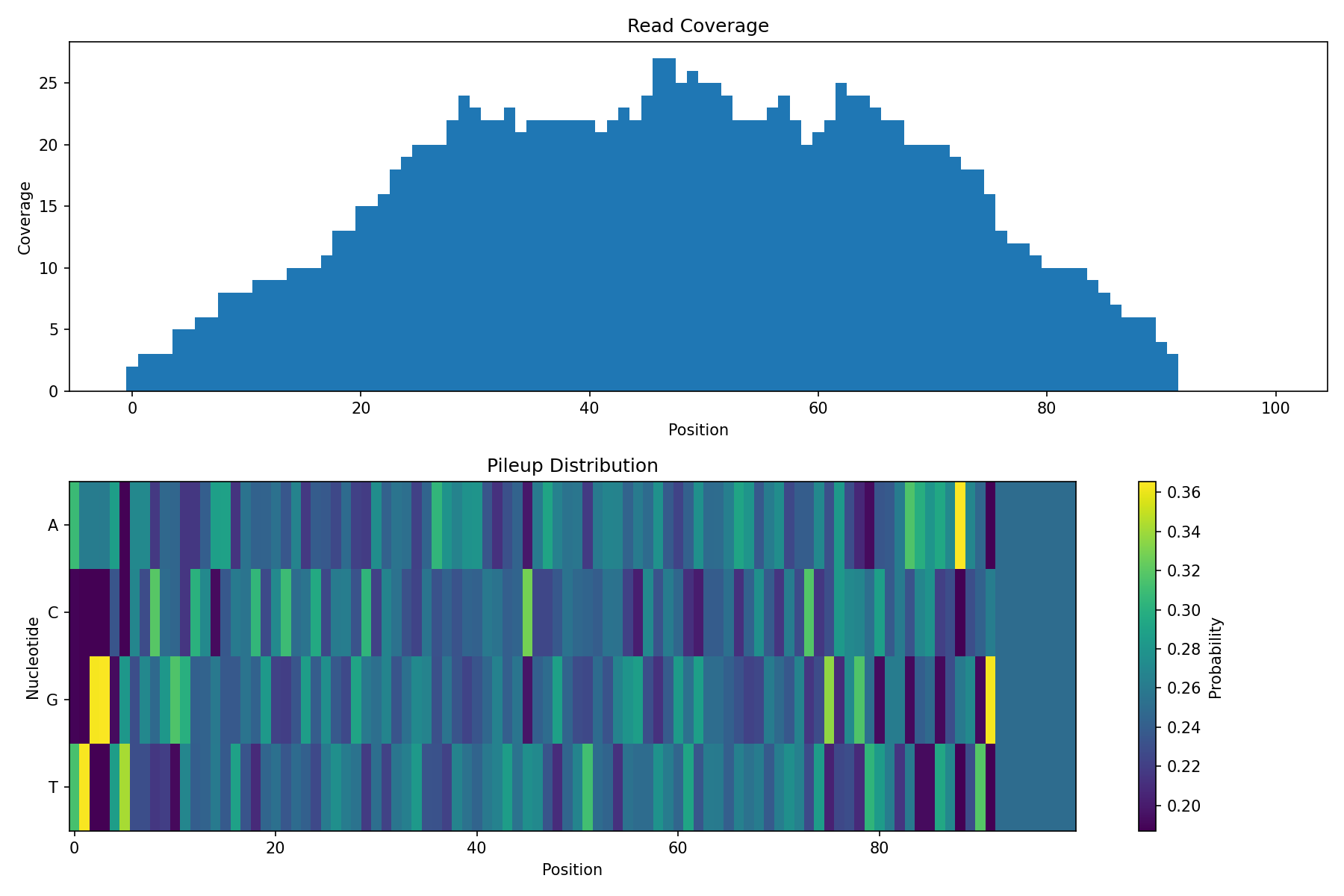
# Visualization
import matplotlib.pyplot as plt
fig, axes = plt.subplots(2, 1, figsize=(12, 8))
# Coverage plot
axes[0].bar(range(reference_length), coverage, width=1.0)
axes[0].set_xlabel('Position')
axes[0].set_ylabel('Coverage')
axes[0].set_title('Read Coverage')
# Pileup heatmap
im = axes[1].imshow(pileup.T, aspect='auto', cmap='viridis')
axes[1].set_xlabel('Position')
axes[1].set_ylabel('Nucleotide')
axes[1].set_yticks([0, 1, 2, 3])
axes[1].set_yticklabels(['A', 'C', 'G', 'T'])
axes[1].set_title('Pileup Distribution')
plt.colorbar(im, ax=axes[1], label='Probability')
plt.tight_layout()
plt.show()
Quality Weighting Effect¤
Compare pileups with and without quality weighting:
# With quality weighting (default)
config_weighted = PileupConfig(
reference_length=100,
use_quality_weights=True,
)
pileup_weighted = DifferentiablePileup(config_weighted)
result_w, _, _ = pileup_weighted.apply(data, {}, None)
# Without quality weighting
config_unweighted = PileupConfig(
reference_length=100,
use_quality_weights=False,
)
pileup_unweighted = DifferentiablePileup(config_unweighted)
result_uw, _, _ = pileup_unweighted.apply(data, {}, None)
# Compare
diff = jnp.abs(result_w["pileup"] - result_uw["pileup"])
print(f"Mean absolute difference: {diff.mean():.4f}")
print(f"Max absolute difference: {diff.max():.4f}")
Output:
Variant Detection¤
Use the pileup to identify potential variants:
# Simulate a reference sequence
key, k_ref = jax.random.split(key)
ref_indices = jax.random.randint(k_ref, (reference_length,), 0, 4)
reference = jax.nn.one_hot(ref_indices, 4)
# Calculate variant likelihood
# Positions where pileup differs from reference
ref_prob = (pileup * reference).sum(axis=-1) # P(reference base)
variant_prob = 1.0 - ref_prob # P(any other base)
# Find high-confidence variants
threshold = 0.3
variant_positions = jnp.where(variant_prob > threshold)[0]
print(f"Potential variants at positions: {variant_positions[:10]}...")
Output:
Gradient Computation¤
The pileup is fully differentiable:
def pileup_loss(reads, positions, quality, target_pileup):
"""Loss function comparing pileup to target."""
data = {"reads": reads, "positions": positions, "quality": quality}
result, _, _ = pileup_op.apply(data, {}, None)
return jnp.mean((result["pileup"] - target_pileup) ** 2)
# Compute gradients w.r.t. reads
grad_fn = jax.grad(pileup_loss)
# Create a target pileup (e.g., all A's)
target = jnp.tile(jnp.array([1.0, 0.0, 0.0, 0.0]), (reference_length, 1))
# Compute gradients
grads = grad_fn(reads, positions, quality, target)
print(f"Gradient shape: {grads.shape}")
print(f"Gradient norm: {jnp.linalg.norm(grads):.4f}")
Output:
Integration with Variant Calling¤
from diffbio.operators import DifferentiableQualityFilter, QualityFilterConfig
# Step 1: Quality filter reads
filter_config = QualityFilterConfig(initial_threshold=20.0)
quality_filter = DifferentiableQualityFilter(filter_config)
# Apply quality filter to each read
filtered_reads = []
for i in range(num_reads):
read_data = {
"sequence": reads[i],
"quality_scores": quality[i],
}
result, _, _ = quality_filter.apply(read_data, {}, None)
filtered_reads.append(result["sequence"])
filtered_reads = jnp.stack(filtered_reads)
# Step 2: Generate pileup from filtered reads
filtered_data = {
"reads": filtered_reads,
"positions": positions,
"quality": quality,
}
result, _, _ = pileup_op.apply(filtered_data, {}, None)
filtered_pileup = result["pileup"]
# Compare filtered vs unfiltered pileup
diff = jnp.abs(pileup - filtered_pileup)
print(f"Effect of quality filtering:")
print(f" Mean change: {diff.mean():.4f}")
print(f" Max change: {diff.max():.4f}")
Output:
Interpreting the Results¤
The quality filtering produces subtle but measurable changes in the pileup:
- Mean change (0.0119): On average, nucleotide probabilities shift by ~1.2% after filtering. This indicates the filter is working but not drastically altering the signal.
- Max change (0.0936): At some positions, probabilities change by up to ~9.4%. These are positions where low-quality bases were significantly down-weighted.
The small mean change with larger max change is expected behavior:
- Most positions have high-quality coverage, so filtering has minimal effect
- Positions with poor-quality reads show larger corrections
- This differential effect is exactly what quality filtering should achieve
Visualizing Quality Filtering Effect¤
# Create full visualization
fig, axes = plt.subplots(2, 2, figsize=(14, 10))
# Top left: Original pileup
im1 = axes[0, 0].imshow(pileup.T, aspect='auto', cmap='viridis', vmin=0, vmax=0.5)
axes[0, 0].set_xlabel('Position')
axes[0, 0].set_ylabel('Nucleotide')
axes[0, 0].set_yticks([0, 1, 2, 3])
axes[0, 0].set_yticklabels(['A', 'C', 'G', 'T'])
axes[0, 0].set_title('Original Pileup (before quality filtering)')
plt.colorbar(im1, ax=axes[0, 0], label='Probability')
# Top right: Filtered pileup
im2 = axes[0, 1].imshow(filtered_pileup.T, aspect='auto', cmap='viridis', vmin=0, vmax=0.5)
axes[0, 1].set_xlabel('Position')
axes[0, 1].set_ylabel('Nucleotide')
axes[0, 1].set_yticks([0, 1, 2, 3])
axes[0, 1].set_yticklabels(['A', 'C', 'G', 'T'])
axes[0, 1].set_title('Filtered Pileup (after quality filtering)')
plt.colorbar(im2, ax=axes[0, 1], label='Probability')
# Bottom left: Difference heatmap
im3 = axes[1, 0].imshow(diff.T, aspect='auto', cmap='Reds', vmin=0, vmax=0.1)
axes[1, 0].set_xlabel('Position')
axes[1, 0].set_ylabel('Nucleotide')
axes[1, 0].set_yticks([0, 1, 2, 3])
axes[1, 0].set_yticklabels(['A', 'C', 'G', 'T'])
axes[1, 0].set_title('Absolute Difference (|original - filtered|)')
plt.colorbar(im3, ax=axes[1, 0], label='Difference')
# Bottom right: Per-position change magnitude
pos_diff = diff.sum(axis=-1) # Sum across nucleotides
axes[1, 1].bar(range(reference_length), pos_diff, width=1.0, color='coral', alpha=0.7)
axes[1, 1].set_xlabel('Position')
axes[1, 1].set_ylabel('Total Change')
axes[1, 1].set_title('Per-Position Quality Filtering Effect')
axes[1, 1].axhline(y=pos_diff.mean(), color='red', linestyle='--', label=f'Mean')
axes[1, 1].legend()
plt.suptitle('Effect of Quality Filtering on Pileup Generation', fontsize=14)
plt.tight_layout()
plt.show()
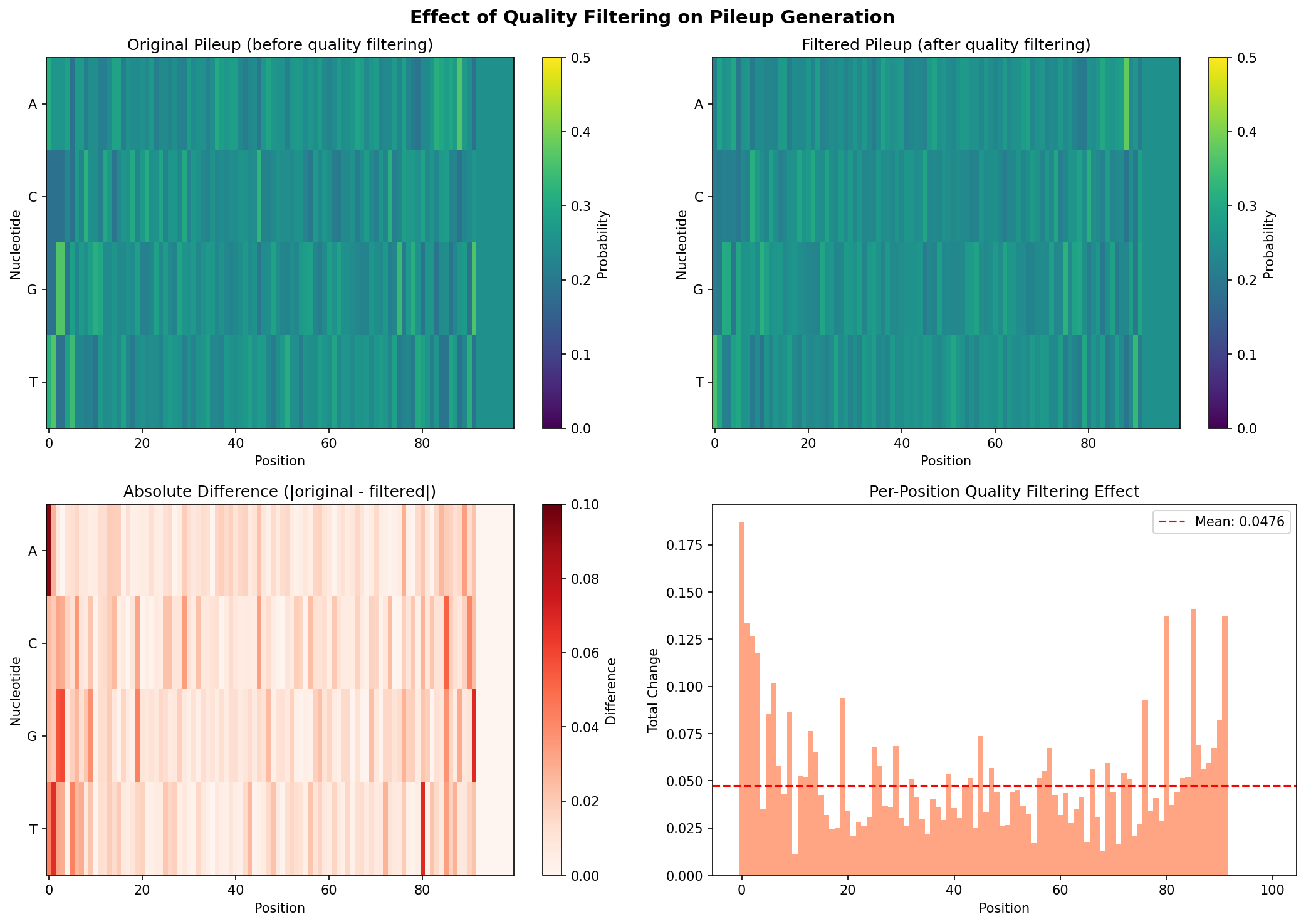
The visualization shows:
-
Top row: Side-by-side comparison of pileup before and after quality filtering. Differences are subtle but visible in regions with lower coverage or quality.
-
Bottom left: A difference heatmap highlighting where the filtering had the most impact. Brighter (red) areas indicate positions where low-quality bases were down-weighted.
-
Bottom right: Per-position total change, showing which genomic positions were most affected by quality filtering. Peaks indicate positions where the quality filter made the largest corrections.
Summary¤
This example demonstrated:
- Creating a differentiable pileup generator
- Preparing read data (reads, positions, quality)
- Generating soft pileups
- Interpreting pileup as nucleotide distributions
- Effect of quality weighting
- Using pileups for variant detection
- Computing gradients through pileup generation
Next Steps¤
- See Variant Calling Pipeline for the complete workflow
- Learn about Training pipelines end-to-end