Molecular Similarity¤
This example demonstrates how to compute molecular similarity using DiffBio's differentiable similarity metrics.
Overview¤
Molecular similarity is fundamental to drug discovery, enabling:
- Virtual screening of compound libraries
- Nearest neighbor search for lead optimization
- Clustering molecules by structural features
DiffBio provides differentiable similarity functions that enable gradient-based optimization of similarity-based objectives.
Prerequisites¤
import jax.numpy as jnp
from flax import nnx
from diffbio.operators.drug_discovery import (
tanimoto_similarity,
cosine_similarity,
dice_similarity,
CircularFingerprintOperator,
CircularFingerprintConfig,
smiles_to_graph,
DEFAULT_ATOM_FEATURES,
)
Basic Similarity Functions¤
Compare two fingerprint vectors using different similarity metrics:
# Create two example fingerprints
fp1 = jnp.array([1.0, 1.0, 0.0, 1.0, 0.0, 0.0, 1.0, 0.0])
fp2 = jnp.array([1.0, 0.0, 1.0, 1.0, 0.0, 1.0, 0.0, 0.0])
print("Fingerprint 1:", [int(x) for x in fp1])
print("Fingerprint 2:", [int(x) for x in fp2])
print(f"\nTanimoto similarity: {float(tanimoto_similarity(fp1, fp2)):.4f}")
print(f"Cosine similarity: {float(cosine_similarity(fp1, fp2)):.4f}")
print(f"Dice similarity: {float(dice_similarity(fp1, fp2)):.4f}")
Output:
Fingerprint 1: [1, 1, 0, 1, 0, 0, 1, 0]
Fingerprint 2: [1, 0, 1, 1, 0, 1, 0, 0]
Tanimoto similarity: 0.3333
Cosine similarity: 0.5000
Dice similarity: 0.5000
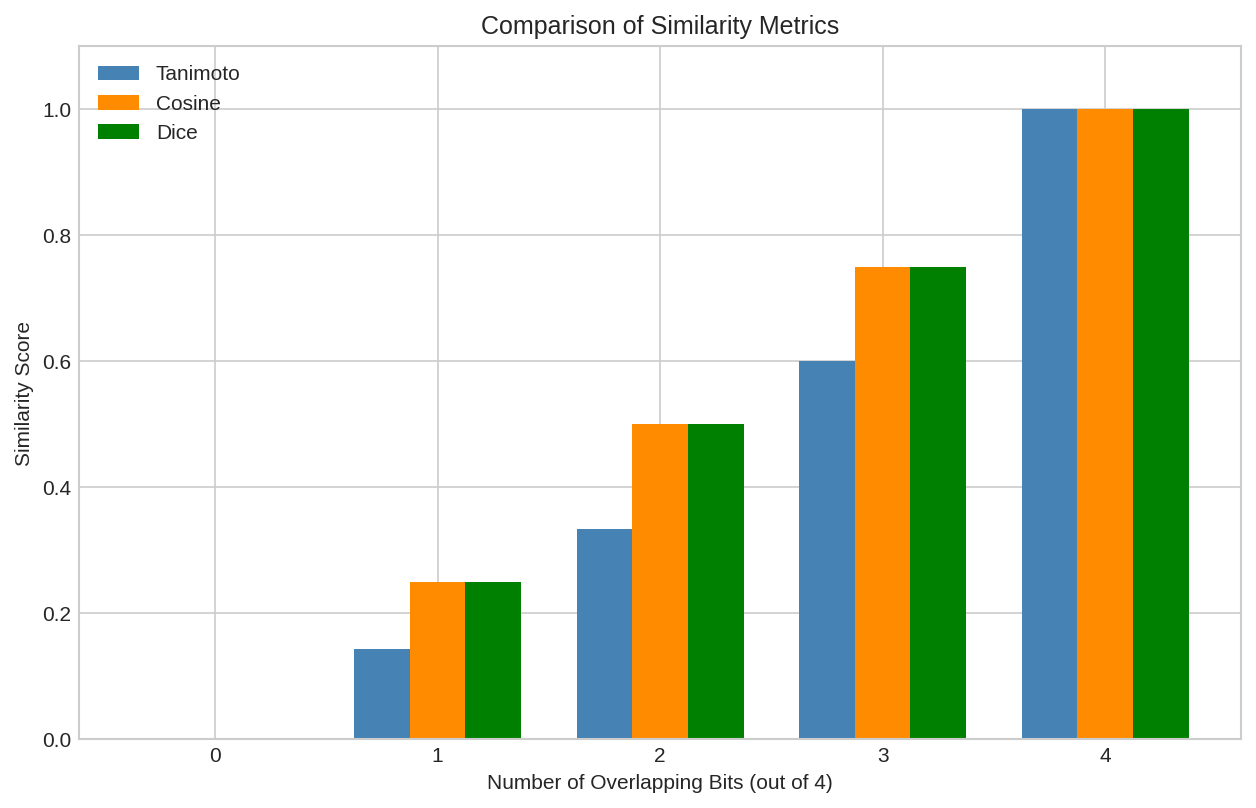
Comparison of Tanimoto, Cosine, and Dice similarity metrics across different fingerprint pairs.
Similarity Metrics Explained¤
Tanimoto Similarity (Jaccard Index)¤
- Most common metric for molecular fingerprints
- Range: 0 (no overlap) to 1 (identical)
- Industry standard for chemical similarity
Cosine Similarity¤
- Measures angle between vectors
- Range: -1 to 1 (0 to 1 for positive fingerprints)
- Good for comparing continuous (neural) fingerprints
Dice Similarity¤
- Related to Tanimoto: \(D = 2T / (1 + T)\)
- More weight to shared features
- Range: 0 to 1
Computing Similarity Between Molecules¤
Compare actual molecules using fingerprints:
# Create fingerprint operator
config = CircularFingerprintConfig(
radius=2,
n_bits=1024,
differentiable=True,
in_features=DEFAULT_ATOM_FEATURES,
)
rngs = nnx.Rngs(42)
fp_op = CircularFingerprintOperator(config, rngs=rngs)
# Generate fingerprints for molecules
molecules = [
("Aspirin", "CC(=O)OC1=CC=CC=C1C(=O)O"),
("Ibuprofen", "CC(C)Cc1ccc(cc1)C(C)C(=O)O"),
("Acetaminophen", "CC(=O)Nc1ccc(O)cc1"),
]
fingerprints = []
for name, smiles in molecules:
graph = smiles_to_graph(smiles)
result, _, _ = fp_op.apply(graph, {}, None)
fingerprints.append(result["fingerprint"])
# Compute pairwise similarities
print("Pairwise Tanimoto Similarities:")
print("-" * 40)
for i, (name_i, _) in enumerate(molecules):
for j, (name_j, _) in enumerate(molecules):
if i < j:
sim = tanimoto_similarity(fingerprints[i], fingerprints[j])
print(f"{name_i} vs {name_j}: {float(sim):.4f}")
Similarity Matrix¤
Build a full similarity matrix for a set of molecules:
import jax.numpy as jnp
def compute_similarity_matrix(fingerprints, similarity_fn=tanimoto_similarity):
"""Compute pairwise similarity matrix."""
n = len(fingerprints)
matrix = jnp.zeros((n, n))
for i in range(n):
for j in range(n):
matrix = matrix.at[i, j].set(
similarity_fn(fingerprints[i], fingerprints[j])
)
return matrix
# Compute similarity matrix
sim_matrix = compute_similarity_matrix(fingerprints)
print("Similarity Matrix:")
print(sim_matrix)
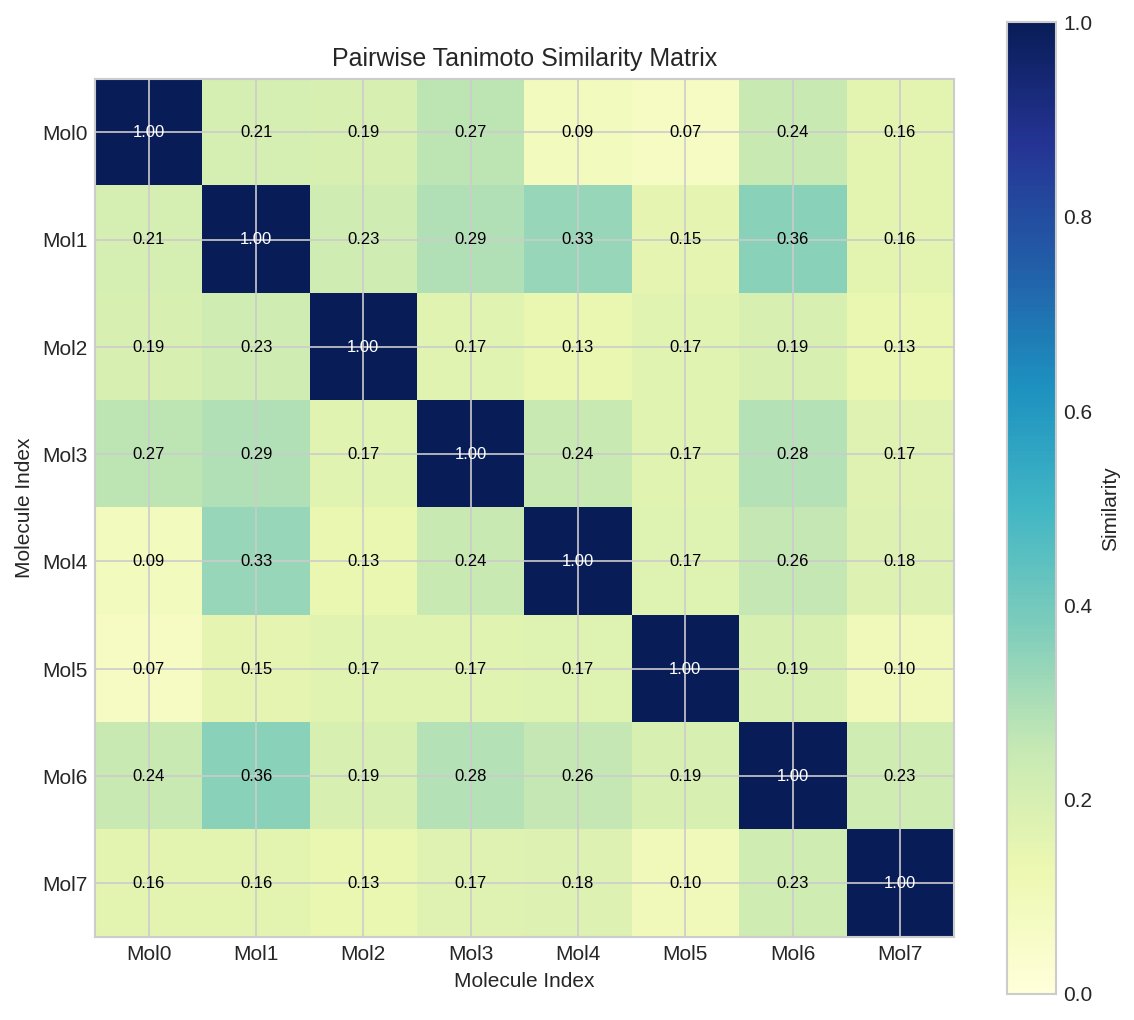
Pairwise Tanimoto similarity matrix for a set of drug molecules. Darker colors indicate higher similarity.
Differentiability¤
All similarity functions are fully differentiable, enabling:
import jax
def similarity_loss(fp1, fp2, target_similarity=0.8):
"""Loss to make two fingerprints more similar."""
current_sim = tanimoto_similarity(fp1, fp2)
return (current_sim - target_similarity) ** 2
# Compute gradient of similarity with respect to fingerprint
grad_fn = jax.grad(lambda fp: float(tanimoto_similarity(fp, fp2)))
grads = grad_fn(fp1)
print(f"Gradient shape: {grads.shape}")
print(f"Gradient norm: {float(jnp.linalg.norm(grads)):.4f}")
Choosing a Similarity Metric¤
| Use Case | Recommended Metric |
|---|---|
| Virtual screening | Tanimoto |
| Binary fingerprints | Tanimoto or Dice |
| Neural fingerprints | Cosine |
| Gradient-based optimization | Any (all differentiable) |
| Asymmetric comparison | Tversky (not shown) |
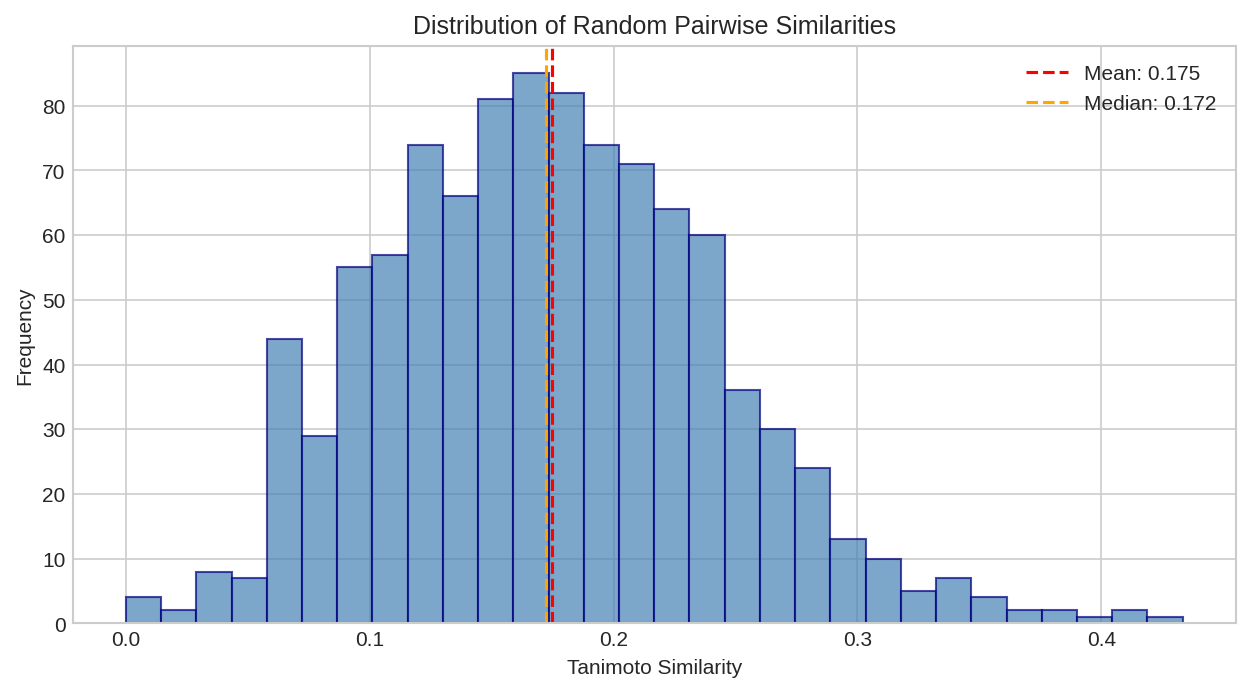
Distribution of similarity scores across a molecular library. Most molecule pairs have low similarity, with a long tail of similar pairs.
Next Steps¤
- Scaffold Splitting - Use Tanimoto clustering for splitting
- Molecular Fingerprints - Generate fingerprints
- Drug Discovery Workflow - Train similarity-based models