HMM Sequence Model¤
This example demonstrates how to use DiffBio's differentiable Hidden Markov Model for sequence analysis.
Overview¤
Hidden Markov Models (HMMs) are powerful tools for:
- Gene finding (coding/non-coding regions)
- Chromatin state annotation
- Profile search (protein domains)
- Sequence labeling
DiffBio implements a fully differentiable HMM using the forward algorithm with logsumexp for numerical stability.
Prerequisites¤
import jax
import jax.numpy as jnp
from flax import nnx
from diffbio.operators.statistical import DifferentiableHMM, HMMConfig
Step 1: Configure the HMM¤
# Create a 3-state HMM for gene finding
# States: intergenic (0), exon (1), intron (2)
# Emissions: DNA bases A=0, C=1, G=2, T=3
config = HMMConfig(
num_states=3, # Hidden states
num_emissions=4, # Observation symbols (DNA bases)
temperature=1.0, # Temperature for softmax
)
rngs = nnx.Rngs(42)
hmm = DifferentiableHMM(config, rngs=rngs)
print(f"Number of states: {config.num_states}")
print(f"Number of emissions: {config.num_emissions}")
Output:
Step 2: Create Observation Sequence¤
# DNA sequence encoded as integers: A=0, C=1, G=2, T=3
observations = jnp.array([0, 1, 2, 3, 0, 1, 2, 3, 0, 0, 1, 1, 2, 2, 3, 3])
print(f"Observation sequence: {observations.tolist()}")
print(f"Sequence length: {len(observations)}")
Output:
Step 3: Compute Likelihood and Posteriors¤
# Apply HMM
data = {"observations": observations}
result, _, _ = hmm.apply(data, {}, None)
log_likelihood = result["log_likelihood"]
posteriors = result["state_posteriors"]
print(f"Log likelihood: {float(log_likelihood):.4f}")
print(f"Posteriors shape: {posteriors.shape}")
Output:
Step 4: Analyze State Posteriors¤
# Show state posteriors for first few positions
print("\nState posteriors (first 5 positions):")
state_names = ["Intergenic", "Exon", "Intron"]
for i in range(5):
probs = posteriors[i]
print(f" Position {i}: ", end="")
for j, name in enumerate(state_names):
print(f"{name[:3]}={float(probs[j]):.3f} ", end="")
print()
Output:
State posteriors (first 5 positions):
Position 0: Int=0.331 Exo=0.371 Intr=0.298
Position 1: Int=0.345 Exo=0.321 Intr=0.334
Position 2: Int=0.326 Exo=0.381 Intr=0.294
Position 3: Int=0.335 Exo=0.349 Intr=0.315
Position 4: Int=0.331 Exo=0.356 Intr=0.313
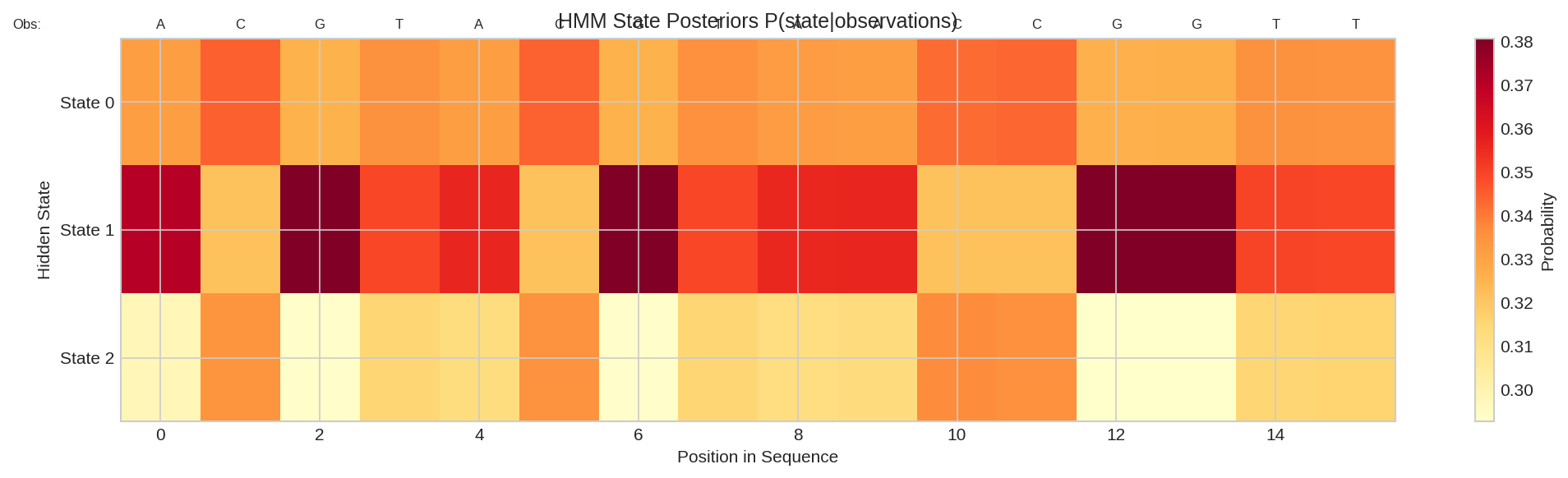
State posteriors P(state|observations) for each position in the sequence. Each column shows the probability distribution over hidden states.
Random Initialization
With random parameters, posteriors are roughly uniform. After training on labeled data, the HMM learns meaningful state assignments.
Understanding the HMM¤
Model Parameters¤
The HMM has three parameter matrices:
- Initial distribution \(\pi\): \(P(\text{state}_0 = i)\)
- Transition matrix \(A\): \(P(\text{state}_t = j | \text{state}_{t-1} = i)\)
- Emission matrix \(B\): \(P(\text{obs}_t = k | \text{state}_t = i)\)
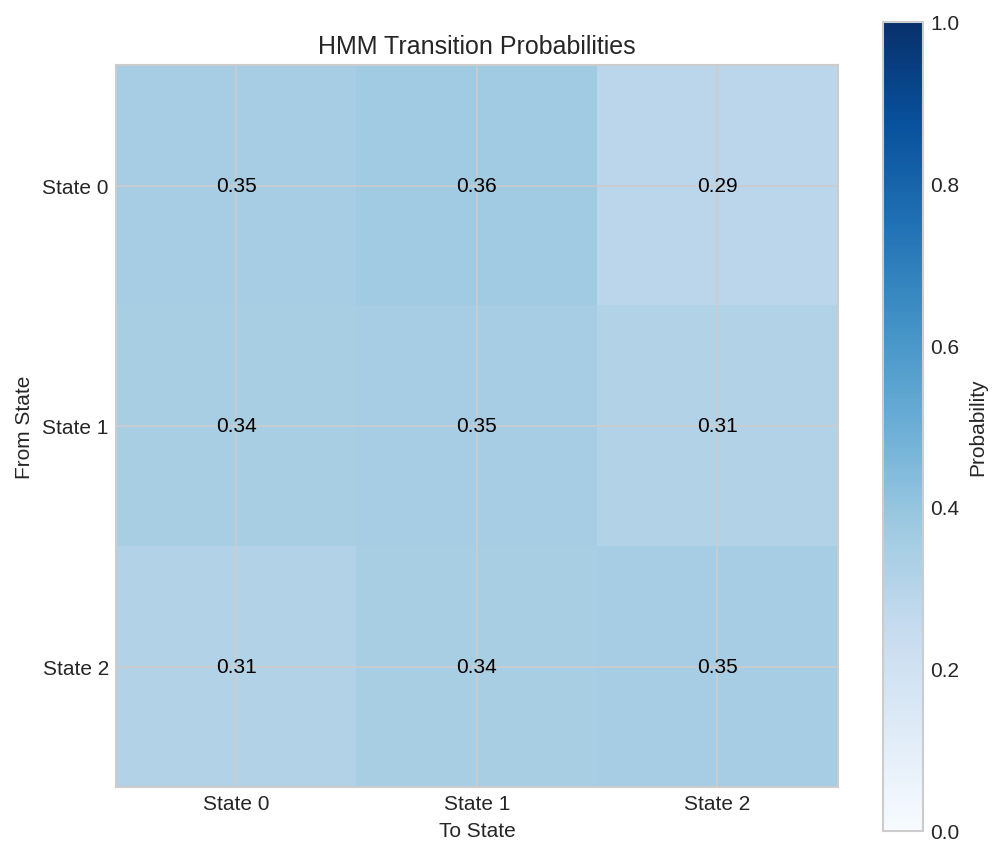
Transition probability matrix showing P(next state | current state). After training, patterns emerge such as high self-transition probability for stable states.
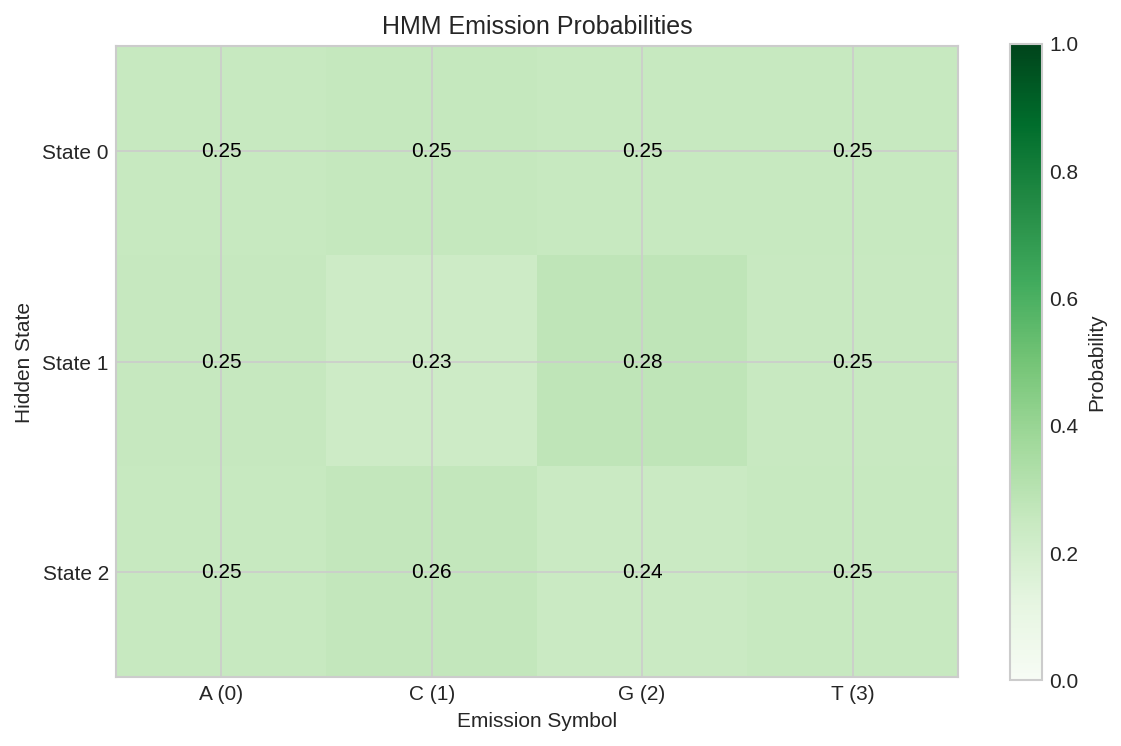
Emission probability matrix showing P(observation | state). Different states learn to emit different DNA base patterns.
Forward Algorithm¤
The forward algorithm computes \(P(\text{observations} | \text{model})\) efficiently:
In log space (for numerical stability): $\(\log \alpha_t(j) = \text{logsumexp}_i(\log \alpha_{t-1}(i) + \log A_{ij}) + \log B_{j,o_t}\)$
State Posteriors¤
The forward-backward algorithm computes:
These posteriors indicate the most likely state at each position.
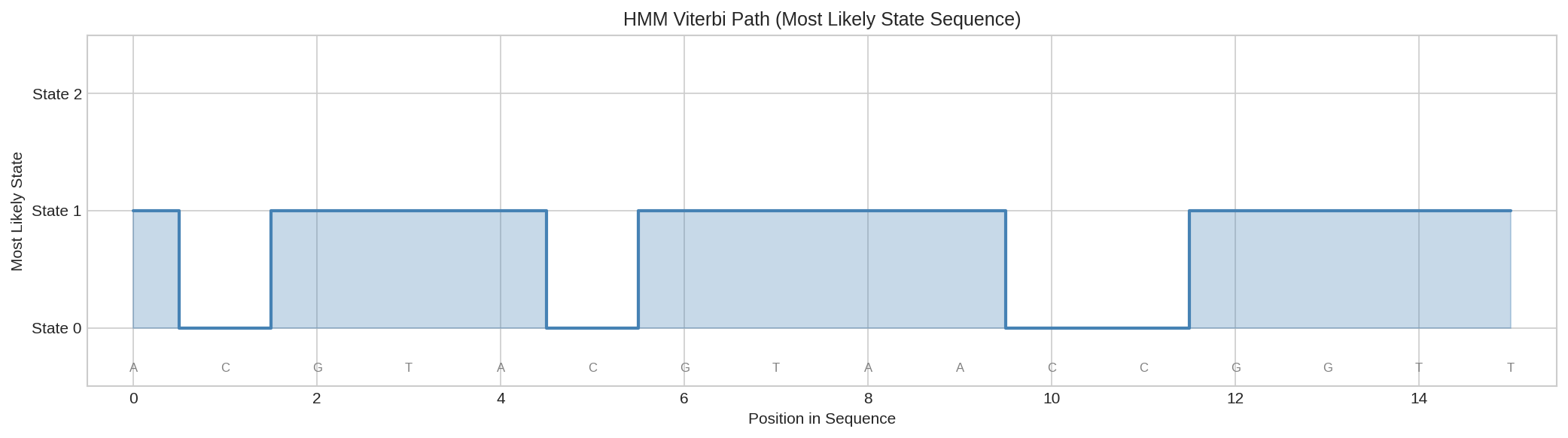
Most likely state sequence (Viterbi path) showing the predicted hidden state at each position based on posterior probabilities.
Differentiability¤
The HMM is fully differentiable, enabling gradient-based training:
def loss_fn(hmm, data):
"""Negative log-likelihood loss."""
result, _, _ = hmm.apply(data, {}, None)
return -result["log_likelihood"]
# Compute gradients
grads = nnx.grad(loss_fn)(hmm, data)
print("Differentiable: Yes (gradient computation successful)")
Output:
Training the HMM¤
Train on labeled sequences (e.g., annotated genes):
import optax
# Create optimizer
optimizer = optax.adam(learning_rate=0.01)
opt_state = optimizer.init(nnx.state(hmm))
# Training step
def train_step(hmm, data, opt_state):
def loss_fn(model):
result, _, _ = model.apply(data, {}, None)
return -result["log_likelihood"]
loss, grads = nnx.value_and_grad(loss_fn)(hmm)
updates, opt_state = optimizer.update(grads, opt_state)
nnx.update(hmm, updates)
return loss, opt_state
# Training loop (pseudocode)
# for epoch in range(epochs):
# for batch in dataloader:
# loss, opt_state = train_step(hmm, batch, opt_state)
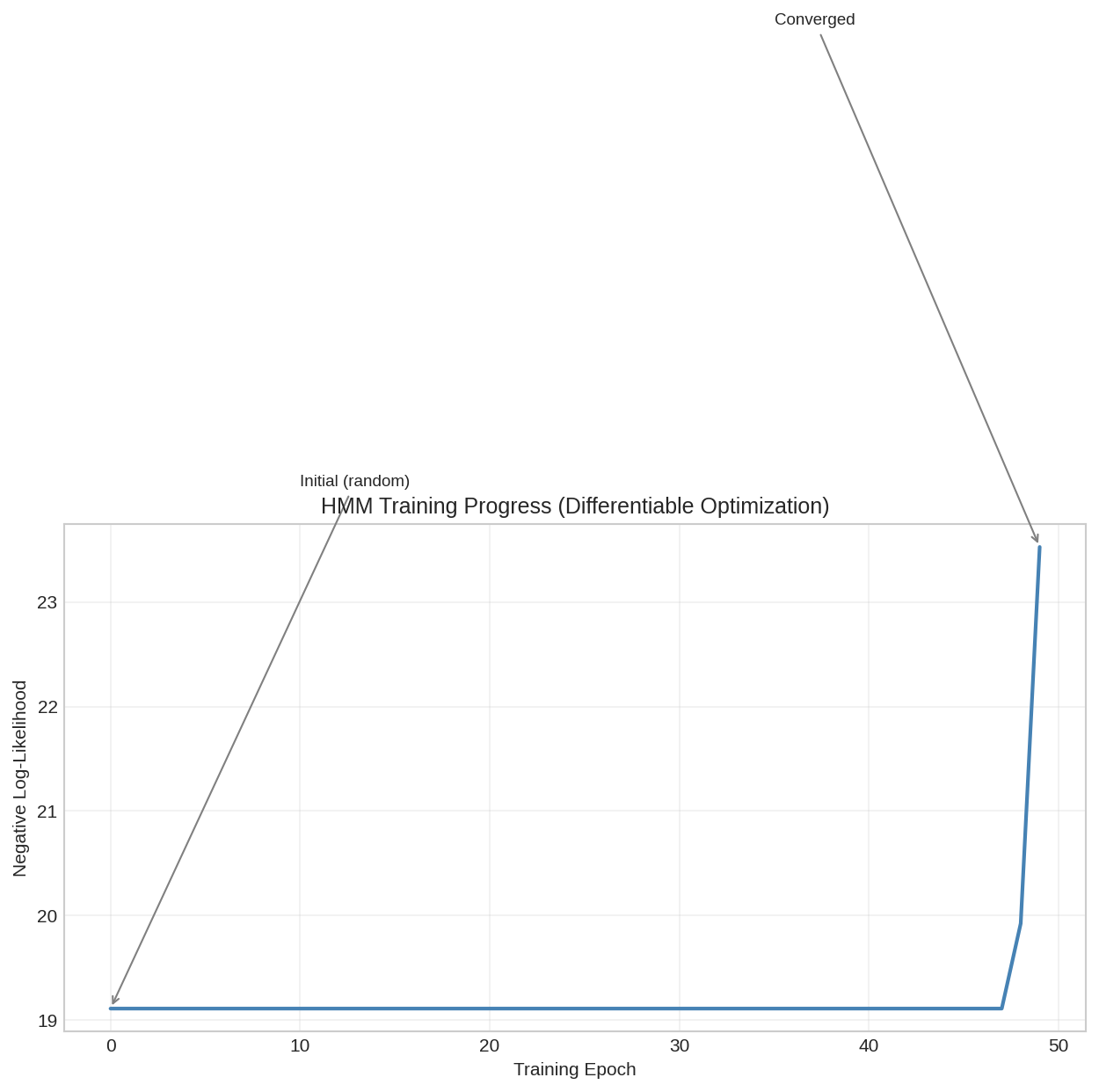
HMM training progress showing negative log-likelihood decreasing over epochs. The differentiable implementation enables gradient-based optimization.
Soft Observations¤
For uncertain observations (e.g., from sequencing), use soft probabilities:
# Soft observations: probability distribution over bases at each position
soft_obs = jnp.array([
[0.9, 0.05, 0.03, 0.02], # Likely A
[0.1, 0.7, 0.1, 0.1], # Likely C
[0.05, 0.05, 0.85, 0.05], # Likely G
# ...
])
# Use forward_soft for soft observations
log_prob = hmm.forward_soft(soft_obs)
Applications¤
| Application | States | Emissions |
|---|---|---|
| Gene finding | Intergenic, Exon, Intron | DNA bases |
| Chromatin states | Active, Poised, Repressed | Histone marks |
| CpG islands | CpG-rich, CpG-poor | Dinucleotides |
| Protein domains | Domain types | Amino acids |
Configuration Options¤
| Parameter | Description | Default |
|---|---|---|
num_states |
Number of hidden states | 3 |
num_emissions |
Number of observation symbols | 4 |
temperature |
Softmax temperature | 1.0 |
learnable_transitions |
Learn transition matrix | True |
learnable_emissions |
Learn emission matrix | True |
Next Steps¤
- DNA Encoding - Encode sequences for HMM
- RNA Structure - Structure-based sequence analysis
- Single-Cell Clustering - Clustering for different domains