DNA Sequence Encoding¤
This example demonstrates how to encode DNA sequences for differentiable bioinformatics operations using DiffBio.
Overview¤
DiffBio represents DNA sequences as one-hot encoded arrays for differentiability. This encoding enables:
- Gradient flow through sequence operations
- Differentiable alignment algorithms
- End-to-end optimization of genomics pipelines
Prerequisites¤
import jax.numpy as jnp
from diffbio.sequences import (
encode_dna_string,
decode_dna_onehot,
gc_content,
reverse_complement_dna,
DNA_ALPHABET,
DNA_ALPHABET_SIZE,
)
DNA Alphabet¤
DiffBio uses the standard DNA alphabet with one-hot encoding:
Output:
The one-hot encoding maps: - A → [1, 0, 0, 0] - C → [0, 1, 0, 0] - G → [0, 0, 1, 0] - T → [0, 0, 0, 1]
Step 1: Encode a DNA Sequence¤
# Example sequence
dna_seq = "ACGTACGT"
print(f"Original sequence: {dna_seq}")
# One-hot encode
encoded = encode_dna_string(dna_seq)
print(f"Encoded shape: {encoded.shape}")
print(f"First position (A): {encoded[0].tolist()}")
print(f"Second position (C): {encoded[1].tolist()}")
Output:
Original sequence: ACGTACGT
Encoded shape: (8, 4)
First position (A): [1.0, 0.0, 0.0, 0.0]
Second position (C): [0.0, 1.0, 0.0, 0.0]
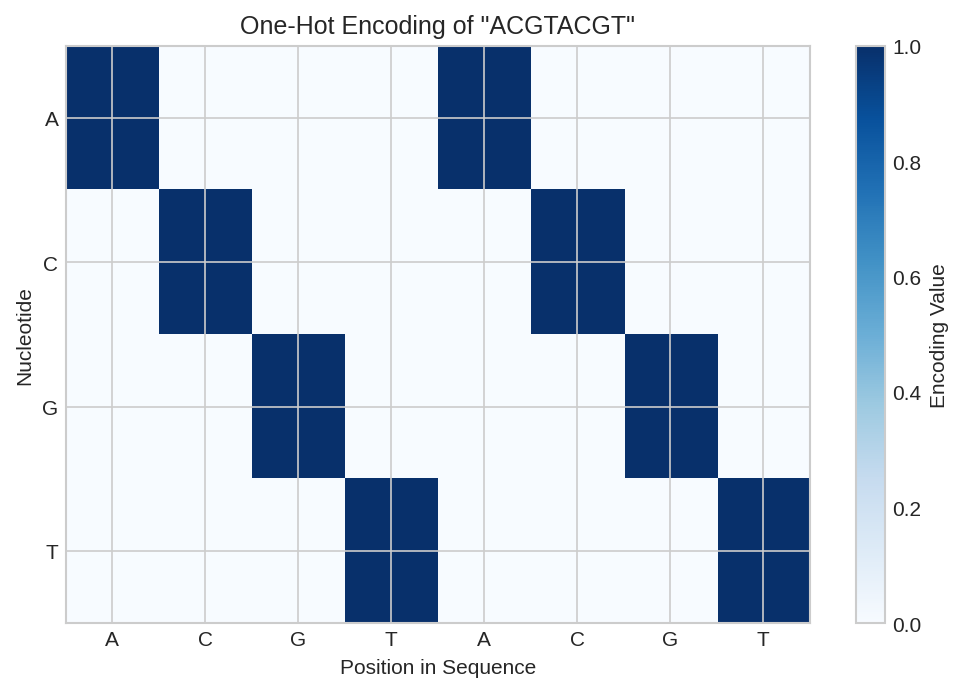
One-hot encoding matrix for DNA sequence "ACGTACGT". Each row represents a position, with the column indicating the base (A=0, C=1, G=2, T=3).
Step 2: Decode Back to String¤
# Decode back to string
decoded = decode_dna_onehot(encoded)
print(f"Decoded back: {decoded}")
assert decoded == dna_seq, "Round-trip encoding works!"
Output:
Step 3: Compute GC Content¤
GC content is the fraction of G and C bases, important for: - Melting temperature prediction - Gene identification - Genome characterization
Output:
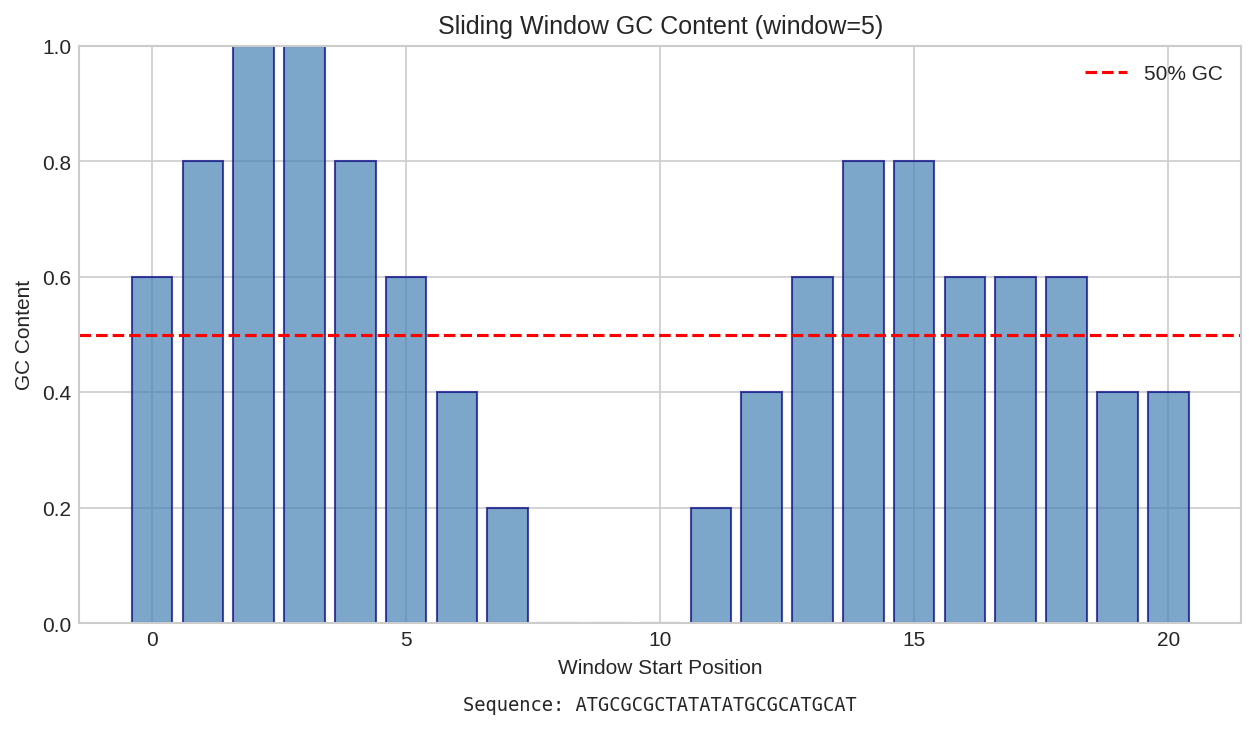
Per-position GC content analysis showing base composition and cumulative GC percentage.
The gc_content function is differentiable, enabling gradient-based optimization of sequence properties.
Step 4: Reverse Complement¤
The reverse complement is essential for working with double-stranded DNA:
# Compute reverse complement
rc = reverse_complement_dna(encoded)
rc_str = decode_dna_onehot(rc)
print(f"Original: {dna_seq}")
print(f"Reverse complement: {rc_str}")
Output:
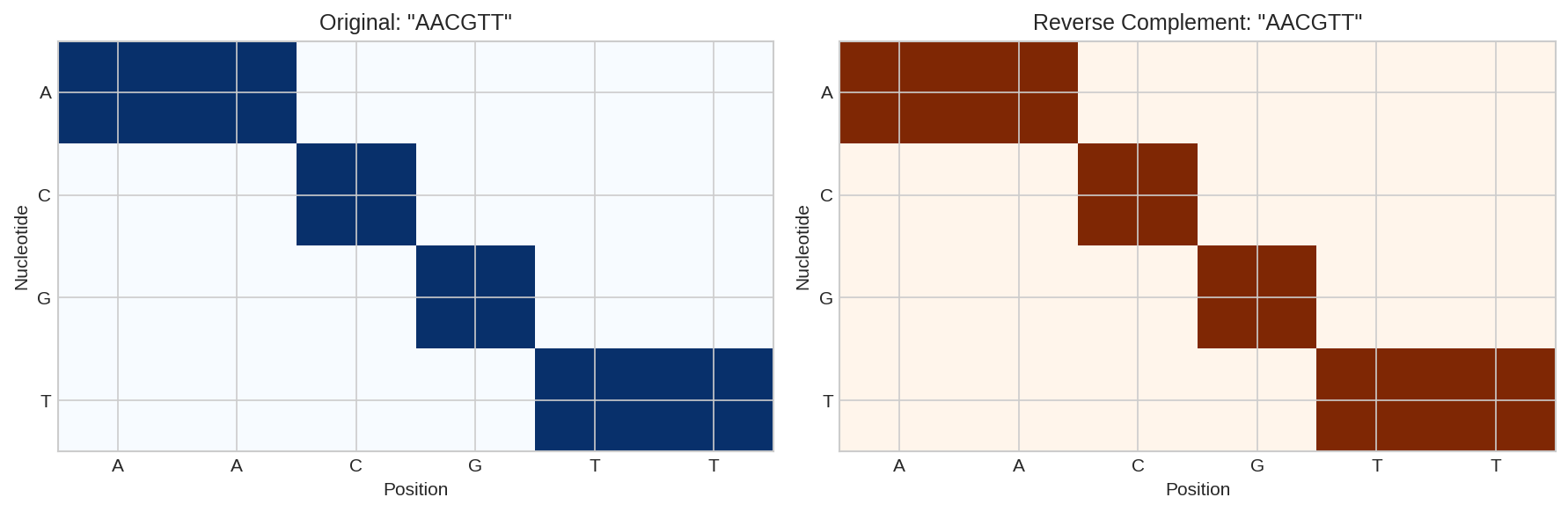
Reverse complement operation showing original sequence and its Watson-Crick complement in reverse orientation.
Palindromic Sequence
The sequence ACGTACGT is palindromic - its reverse complement equals itself. This is common in restriction enzyme recognition sites.
Soft Encoding for Uncertainty¤
For sequences with uncertainty (e.g., from sequencing errors), use soft encoding:
from diffbio.sequences import soft_encode_dna
# Soft encode with quality scores
sequence = "ACGT"
qualities = jnp.array([30, 20, 10, 5]) # Phred scores
soft_encoded = soft_encode_dna(sequence, qualities)
print("Soft encoded probabilities:")
for i, base in enumerate(sequence):
probs = soft_encoded[i]
print(f" Position {i} ({base}): {[f'{p:.3f}' for p in probs]}")
Lower quality scores spread probability across all bases, reflecting uncertainty.
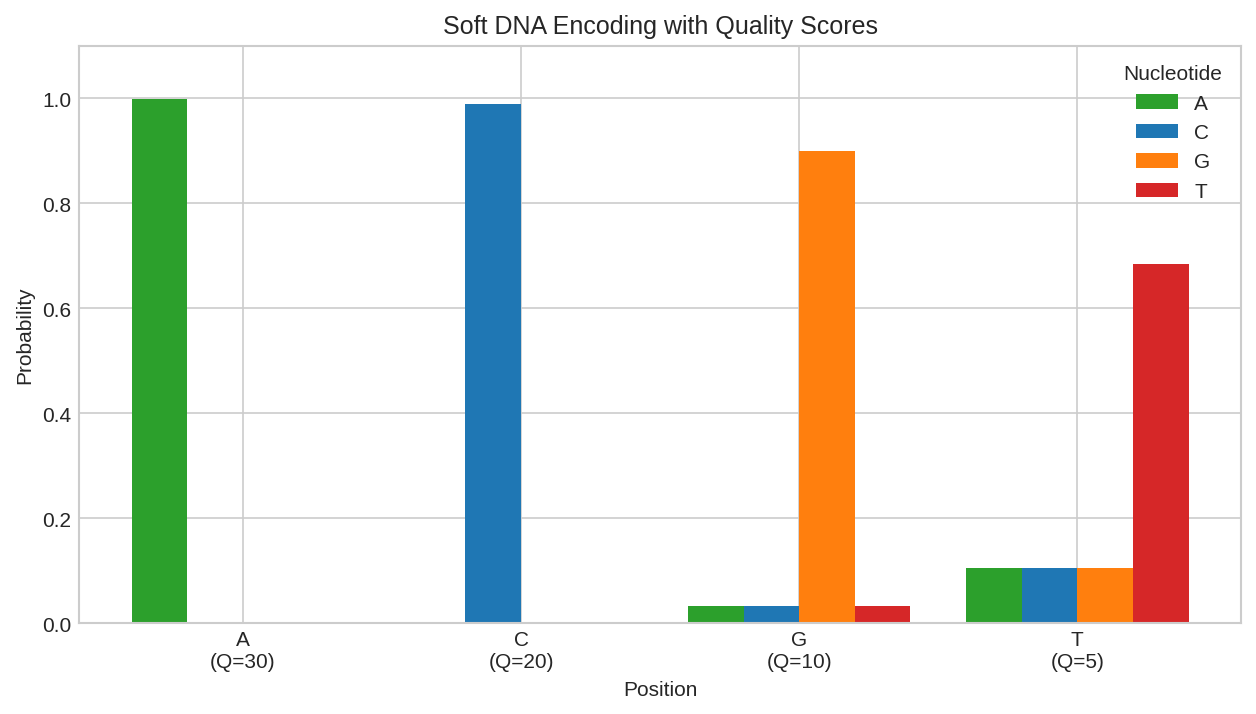
Soft encoding with quality scores: higher quality positions have sharper distributions, while lower quality positions spread probability across bases.
Using with Alignment¤
One-hot encoded sequences integrate directly with DiffBio's alignment operators:
from diffbio.operators.alignment import (
SmoothSmithWaterman,
SmithWatermanConfig,
create_dna_scoring_matrix,
)
from flax import nnx
# Create sequences
seq1 = encode_dna_string("ACGTACGT")
seq2 = encode_dna_string("ACGTTACGT")
# Create aligner
scoring_matrix = create_dna_scoring_matrix(match=2.0, mismatch=-1.0)
config = SmithWatermanConfig(temperature=1.0, gap_open=-2.0, gap_extend=-0.5)
aligner = SmoothSmithWaterman(config, scoring_matrix=scoring_matrix, rngs=nnx.Rngs(42))
# Align
data = {"seq1": seq1, "seq2": seq2}
result, _, _ = aligner.apply(data, {}, None)
print(f"Alignment score: {float(result['score']):.2f}")
Output:
Differentiability¤
All sequence operations are differentiable:
import jax
def sequence_loss(encoded):
"""Example loss: minimize GC content."""
return gc_content(encoded)
# Compute gradient
grad_fn = jax.grad(sequence_loss)
grads = grad_fn(encoded)
print(f"Gradient shape: {grads.shape}")
print(f"Gradient at position 0 (A): {grads[0].tolist()}")
Gradients indicate how changing each position affects the loss, enabling sequence optimization.
Summary¤
| Function | Purpose | Differentiable |
|---|---|---|
encode_dna_string |
String → one-hot | N/A (input) |
decode_dna_onehot |
One-hot → string | N/A (output) |
gc_content |
Compute GC fraction | Yes |
reverse_complement_dna |
Reverse complement | Yes |
soft_encode_dna |
Quality-aware encoding | Yes |
Next Steps¤
- Sequence Alignment - Align sequences using Smith-Waterman
- Variant Calling Pipeline - End-to-end genomics
- HMM Sequence Model - Hidden Markov Models for sequences