Scaffold Splitting¤
This example demonstrates how to properly split molecular datasets using scaffold-based splitting for drug discovery benchmarks.
Overview¤
In drug discovery, random data splitting leads to data leakage because structurally similar molecules end up in both training and test sets. Scaffold splitting ensures:
- Molecules with the same core scaffold are in the same split
- Test molecules have different scaffolds than training molecules
- More realistic evaluation of generalization to new chemical series
Prerequisites¤
from diffbio.sources import MolNetSource, MolNetSourceConfig
from diffbio.splitters import ScaffoldSplitter, ScaffoldSplitterConfig
What is a Molecular Scaffold?¤
A Bemis-Murcko scaffold is the core ring structure of a molecule after removing side chains:
Molecules sharing the same scaffold often have similar properties, so putting them in different splits creates optimistic performance estimates.
Step 1: Load Dataset¤
# Load BBBP dataset
source_config = MolNetSourceConfig(
dataset_name="bbbp",
split="train"
)
source = MolNetSource(source_config)
print(f"Dataset size: {len(source)}")
Output:
Step 2: Create Scaffold Splitter¤
# Configure scaffold-based splitting
split_config = ScaffoldSplitterConfig(
train_frac=0.8,
valid_frac=0.1,
test_frac=0.1,
)
splitter = ScaffoldSplitter(split_config)
# Perform the split
result = splitter.split(source)
print(f"Train size: {len(result.train_indices)} ({len(result.train_indices)/len(source):.1%})")
print(f"Valid size: {len(result.valid_indices)} ({len(result.valid_indices)/len(source):.1%})")
print(f"Test size: {len(result.test_indices)} ({len(result.test_indices)/len(source):.1%})")
Output:
Step 3: Verify Scaffold Separation¤
Count unique scaffolds in each split to verify proper separation:
from rdkit import Chem
from rdkit.Chem.Scaffolds import MurckoScaffold
def count_unique_scaffolds(indices, source, max_count=100):
"""Count unique scaffolds in a split."""
scaffolds = set()
for idx in indices[:max_count]:
smiles = source[int(idx)].data["smiles"]
mol = Chem.MolFromSmiles(smiles)
if mol:
try:
scaffold = MurckoScaffold.MurckoScaffoldSmiles(mol=mol)
scaffolds.add(scaffold)
except Exception:
pass
return len(scaffolds)
train_scaffolds = count_unique_scaffolds(result.train_indices, source)
test_scaffolds = count_unique_scaffolds(result.test_indices, source)
print(f"\nUnique scaffolds (first 100):")
print(f" Train: {train_scaffolds}")
print(f" Test: {test_scaffolds}")
Output:
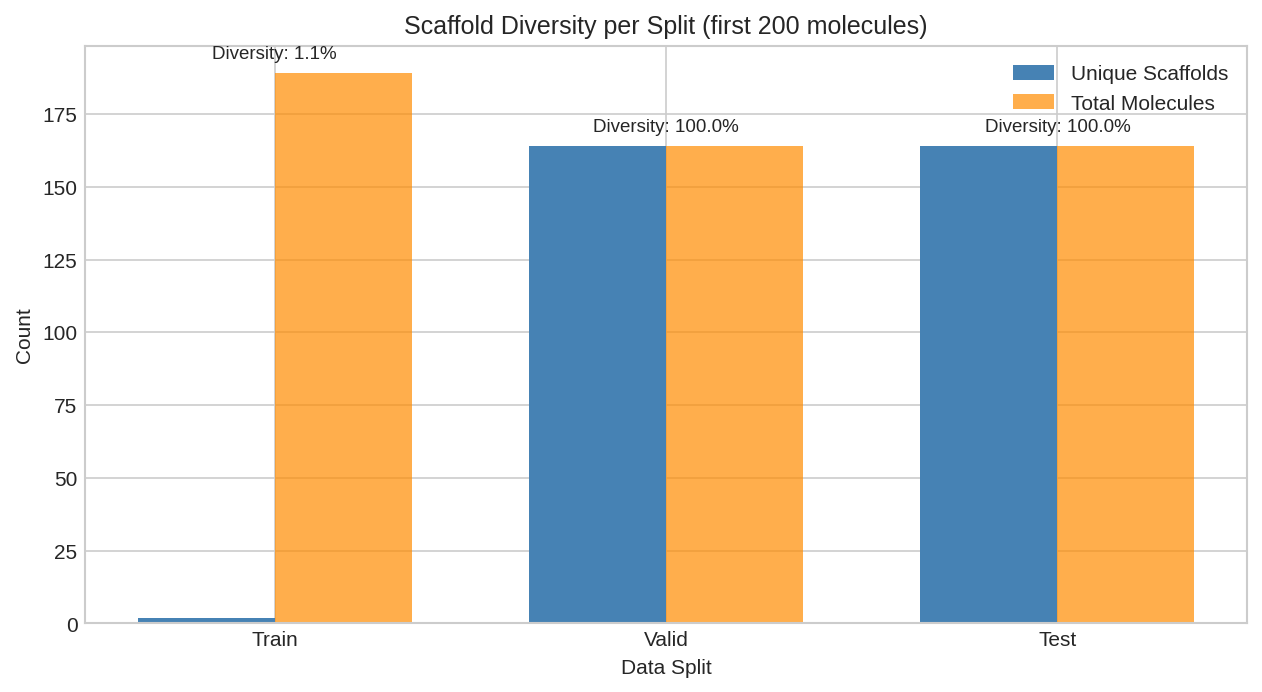
Number of unique scaffolds in each split. Training contains fewer scaffolds with more molecules per scaffold, while test has many unique scaffolds.
Scaffold Distribution
Largest scaffolds go to training first. The train set often has fewer unique scaffolds but more molecules per scaffold, while test has many unique scaffolds with fewer molecules each.
Using Split Indices¤
Access molecules by split:
from diffbio.sources import IndexedViewSource
# Create views for each split
train_view = IndexedViewSource(source, result.train_indices)
valid_view = IndexedViewSource(source, result.valid_indices)
test_view = IndexedViewSource(source, result.test_indices)
print(f"Training molecules: {len(train_view)}")
print(f"Validation molecules: {len(valid_view)}")
print(f"Test molecules: {len(test_view)}")
# Access elements
train_element = train_view[0]
print(f"\nFirst training molecule: {train_element.data['smiles'][:50]}...")
Alternative: Tanimoto Cluster Splitting¤
For finer control, use fingerprint similarity clustering:
from diffbio.splitters import TanimotoClusterSplitter, TanimotoClusterSplitterConfig
config = TanimotoClusterSplitterConfig(
train_frac=0.8,
valid_frac=0.1,
test_frac=0.1,
fingerprint_type="morgan",
fingerprint_radius=2,
similarity_cutoff=0.6, # Cluster threshold
)
splitter = TanimotoClusterSplitter(config)
result = splitter.split(source)
Comparison: Random vs Scaffold Splitting¤
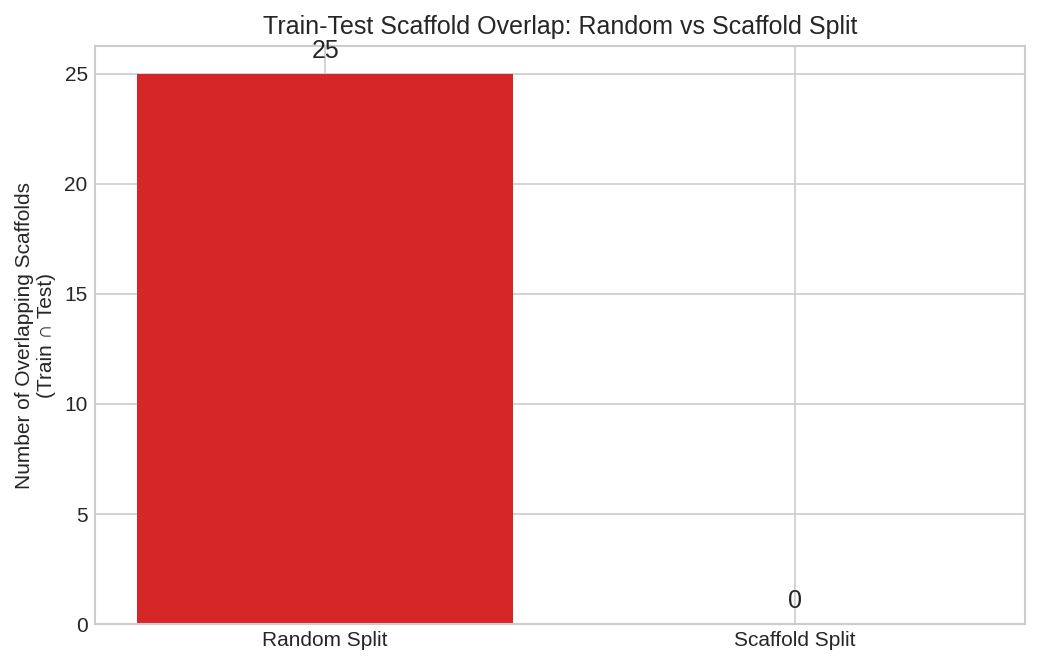
Comparison of random vs scaffold splitting showing scaffold overlap between splits. Random splitting allows similar molecules in train and test, inflating performance estimates.
| Aspect | Random Split | Scaffold Split |
|---|---|---|
| Implementation | Simple | Requires RDKit |
| Similar molecules in same split? | No guarantee | Yes (by design) |
| Realistic evaluation | No | Yes |
| Performance estimate | Optimistic | More realistic |
| Use case | Quick experiments | Publication-ready results |
Example molecules and their Bemis-Murcko scaffolds. Molecules with the same scaffold are grouped together in scaffold splitting.
Best Practices¤
-
Always use scaffold splitting for drug discovery benchmarks - Random splitting inflates performance metrics
-
Report both splits - If comparing to literature, report performance on both random and scaffold splits
-
Use consistent seed - For reproducibility, use the same random seed for splitting
-
Validate on different scaffolds - Ensure validation set also has distinct scaffolds from training
Next Steps¤
- MolNet Data Loading - Load benchmark datasets
- Molecular Fingerprints - Generate fingerprints for clustering
- Drug Discovery Workflow - Train with proper splitting