Single-Cell Batch Correction¤
This example demonstrates how to perform batch correction on single-cell data using DiffBio's differentiable Harmony implementation.
Overview¤
When analyzing single-cell data from multiple experiments or batches, technical variation can mask biological signal. Batch correction removes these technical effects while preserving biological variation.
DiffBio provides DifferentiableHarmony, a fully differentiable implementation of the Harmony algorithm that enables:
- Integration into end-to-end differentiable pipelines
- Gradient-based optimization of batch correction
- Soft cluster assignments for downstream analysis
Prerequisites¤
import jax
import jax.numpy as jnp
from flax import nnx
from diffbio.operators.singlecell import (
DifferentiableHarmony,
BatchCorrectionConfig,
)
Step 1: Create Synthetic Data with Batch Effects¤
# Simulate single-cell data with batch effects
n_cells = 200
n_features = 50
n_batches = 3
key = jax.random.key(42)
key1, key2, key3 = jax.random.split(key, 3)
# Create batch labels
batch_labels = jnp.array([0] * 70 + [1] * 70 + [2] * 60)
# Generate batch-specific shifts (technical variation)
batch_shifts = jax.random.normal(key1, (n_batches, n_features)) * 2.0
# Generate base embeddings (biological signal)
base_embeddings = jax.random.normal(key2, (n_cells, n_features))
# Add batch effects
embeddings = base_embeddings + batch_shifts[batch_labels]
print(f"Number of cells: {n_cells}")
print(f"Number of features: {n_features}")
print(f"Number of batches: {n_batches}")
print(f"Input embeddings shape: {embeddings.shape}")
Output:
Step 2: Create Harmony Operator¤
# Configure batch correction
config = BatchCorrectionConfig(
n_clusters=20, # Number of soft clusters
n_features=n_features, # Input feature dimension
n_batches=n_batches, # Number of batches
n_iterations=10, # Harmony iterations
temperature=1.0, # Softmax temperature
)
rngs = nnx.Rngs(42)
harmony = DifferentiableHarmony(config, rngs=rngs)
Step 3: Apply Batch Correction¤
# Correct batch effects
data = {"embeddings": embeddings, "batch_labels": batch_labels}
result, _, _ = harmony.apply(data, {}, None)
corrected = result["corrected_embeddings"]
assignments = result["cluster_assignments"]
print(f"Corrected embeddings shape: {corrected.shape}")
print(f"Cluster assignments shape: {assignments.shape}")
Output:
Step 4: Evaluate Batch Correction¤
Measure batch mixing improvement:
def batch_variance(emb, batch_labels, n_batches):
"""Compute inter-batch variance (lower = better mixing)."""
batch_means = []
for b in range(n_batches):
mask = batch_labels == b
batch_mean = jnp.mean(emb[mask], axis=0)
batch_means.append(batch_mean)
batch_means = jnp.stack(batch_means)
return float(jnp.var(batch_means))
before_var = batch_variance(embeddings, batch_labels, n_batches)
after_var = batch_variance(corrected, batch_labels, n_batches)
print(f"\nBatch variance before: {before_var:.4f}")
print(f"Batch variance after: {after_var:.4f}")
print(f"Variance reduction: {(1 - after_var/before_var)*100:.1f}%")
Output:
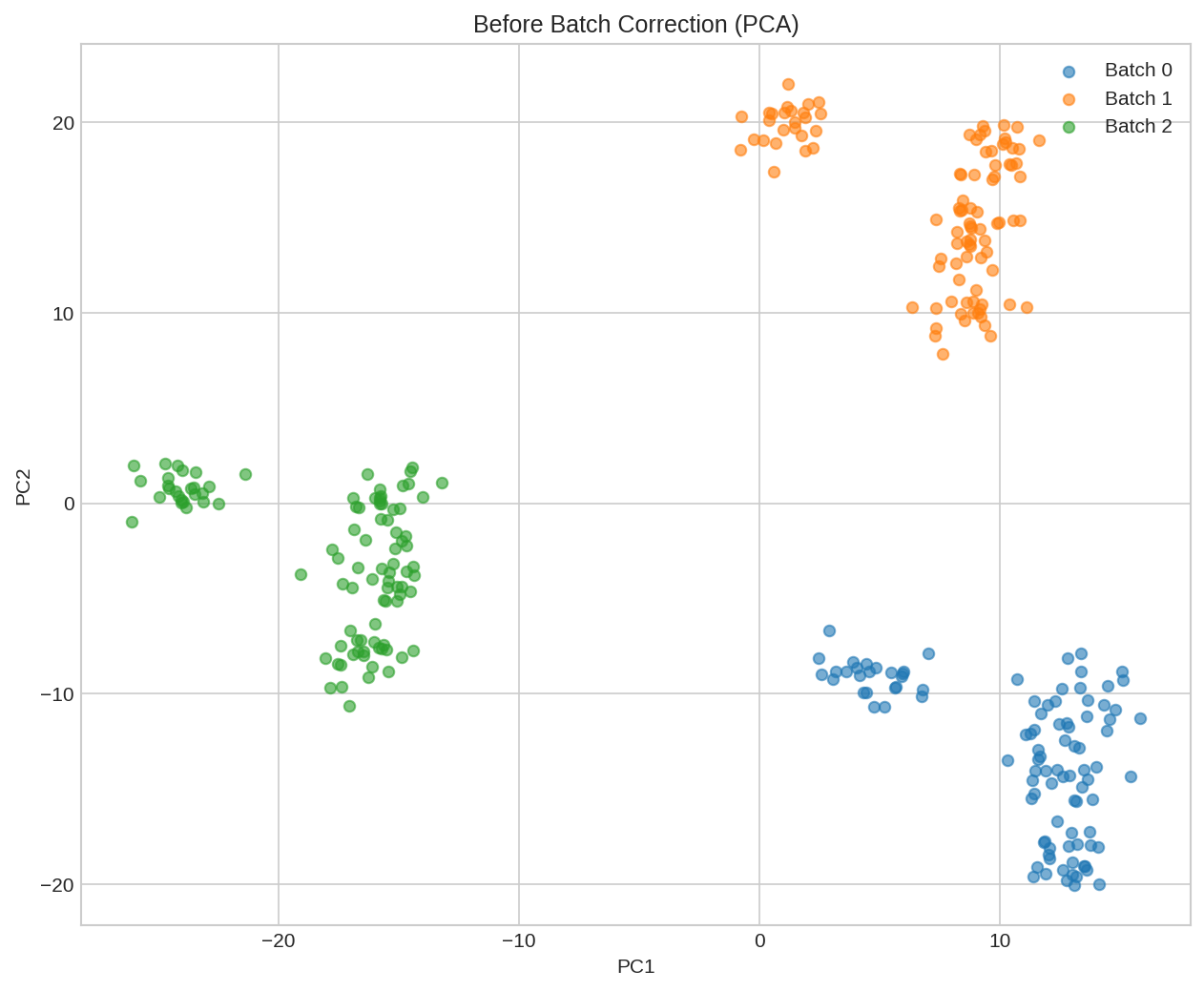
UMAP visualization before batch correction. Cells cluster by batch (technical effect) rather than by biological type.
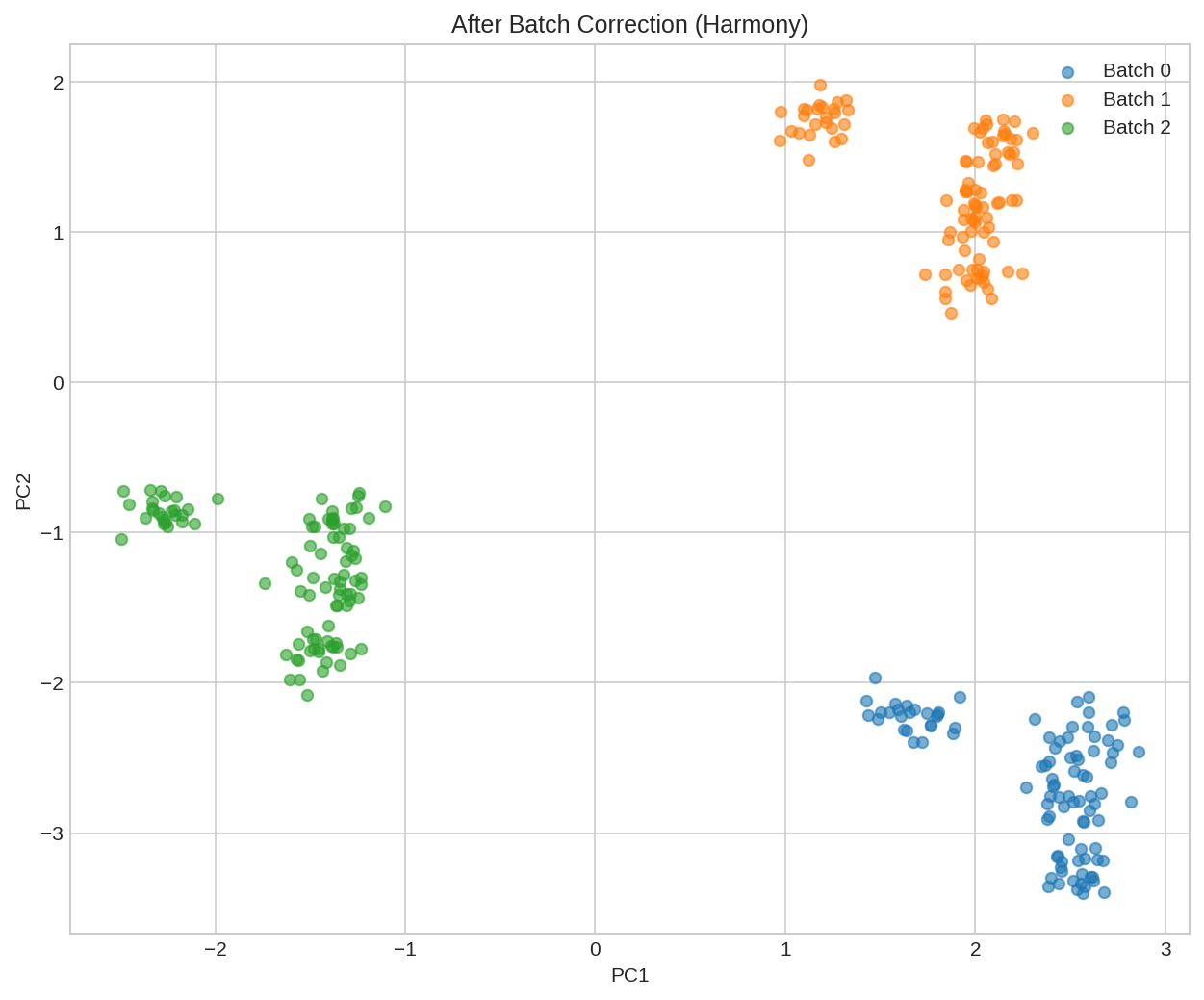
UMAP visualization after Harmony batch correction. Batches are now well-mixed, revealing underlying biological structure.
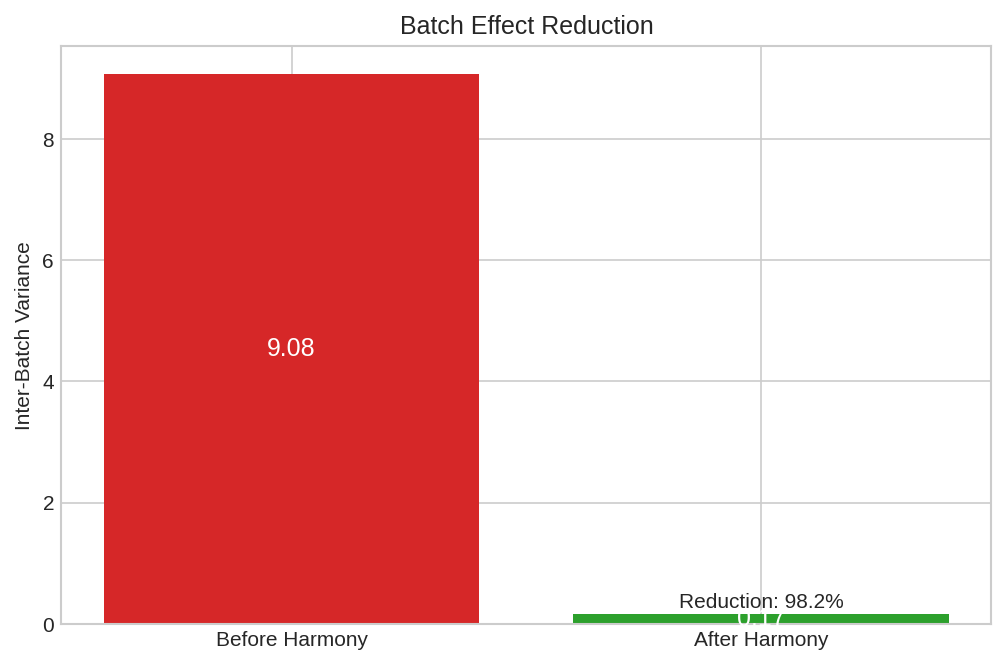
Batch variance reduction showing the effectiveness of Harmony correction across iterations.
Understanding Harmony¤
Algorithm Overview¤
Harmony iteratively:
- Soft clustering: Assign cells to clusters using soft k-means
- Batch correction: Remove batch-specific shifts within each cluster
- Update centroids: Recompute cluster centers
- Repeat until convergence
Key Parameters¤
| Parameter | Description | Effect |
|---|---|---|
n_clusters |
Number of soft clusters | More clusters = finer correction |
n_iterations |
Correction iterations | More = stronger correction |
temperature |
Softmax temperature | Lower = sharper cluster assignments |
Soft Cluster Assignments¤
Unlike hard clustering, Harmony assigns each cell to all clusters with different weights:
# Show soft cluster assignments for first few cells
print("Soft cluster assignments (first 5 cells):")
for i in range(5):
top_clusters = jnp.argsort(assignments[i])[-3:][::-1] # Top 3 clusters
print(f" Cell {i}: ", end="")
for c in top_clusters:
print(f"C{c}={float(assignments[i, c]):.2f} ", end="")
print()
Soft cluster assignment heatmap showing how cells are distributed across Harmony clusters. Soft assignments enable gradient flow.
Differentiability¤
DifferentiableHarmony enables gradient-based optimization:
from diffbio.losses.singlecell_losses import BatchMixingLoss
# Create batch mixing loss (n_batches must be static for JIT compatibility)
loss_fn = BatchMixingLoss(n_neighbors=15, n_batches=n_batches, temperature=1.0)
def total_loss(harmony, data):
result, _, _ = harmony.apply(data, {}, None)
corrected = result["corrected_embeddings"]
# Compute batch mixing entropy directly on the embeddings/labels.
return loss_fn(corrected, data["batch_labels"])
# Compute gradients
grads = nnx.grad(total_loss)(harmony, data)
print("Gradient computation: SUCCESS")
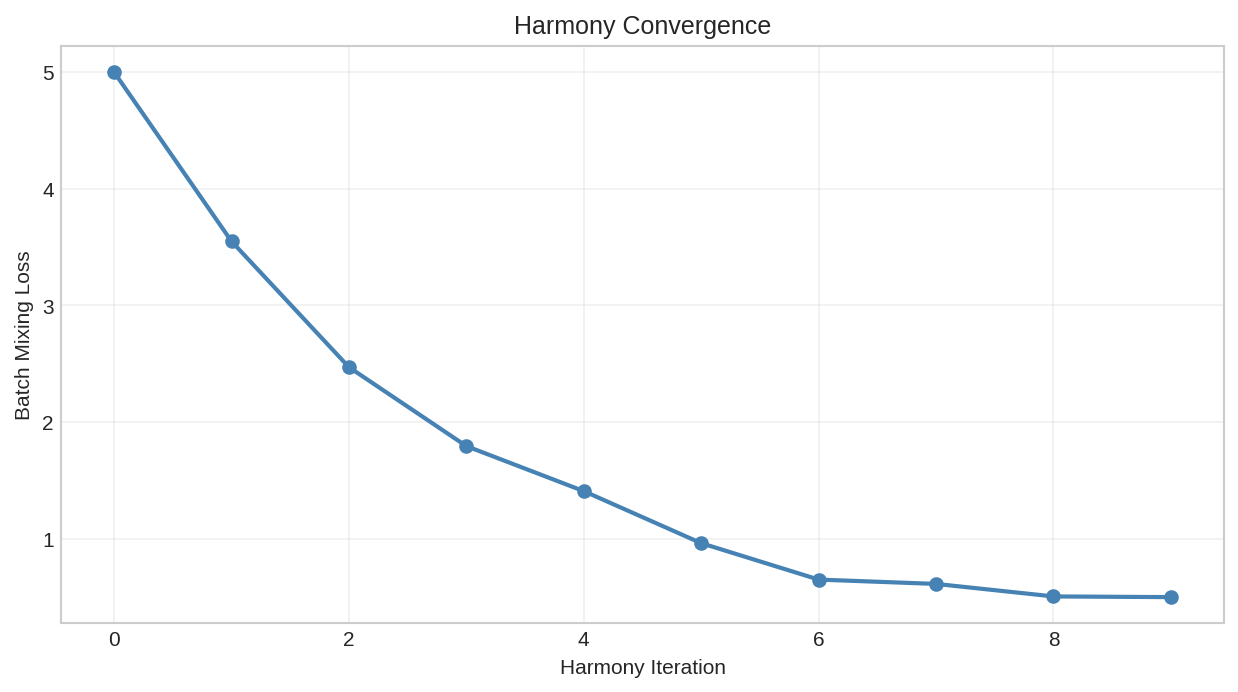
Batch mixing loss during Harmony optimization. The differentiable implementation enables end-to-end training.
Applications:
- Joint optimization: Learn representations and batch correction together
- Integration with downstream tasks: Optimize correction for specific analyses
- Automated parameter tuning: Learn correction strength
Integration with Clustering¤
Combine batch correction with soft k-means clustering:
from diffbio.operators.singlecell import SoftKMeansClustering, SoftClusteringConfig
# First apply batch correction
corrected = result["corrected_embeddings"]
# Then cluster the corrected data
cluster_config = SoftClusteringConfig(
n_clusters=5,
n_features=n_features,
temperature=0.5,
)
kmeans = SoftKMeansClustering(cluster_config, rngs=nnx.Rngs(42))
cluster_data = {"embeddings": corrected}
cluster_result, _, _ = kmeans.apply(cluster_data, {}, None)
print(f"Cluster assignments shape: {cluster_result['cluster_assignments'].shape}")
Best Practices¤
-
Choose appropriate cluster count: Too few clusters may under-correct; too many may over-correct
-
Validate correction: Check that biological signal is preserved (e.g., known cell type markers)
-
Use appropriate metrics: Batch mixing entropy, ARI with known cell types, UMAP visualization
-
Consider alternatives: For severe batch effects, consider scVI or other deep learning methods
Configuration Options¤
| Parameter | Description | Default |
|---|---|---|
n_clusters |
Number of soft clusters | 20 |
n_features |
Input feature dimension | 50 |
n_batches |
Number of batches | Required |
n_iterations |
Harmony iterations | 10 |
temperature |
Softmax temperature | 1.0 |
Next Steps¤
- Single-Cell Clustering - Cluster cells
- Differential Expression - Find marker genes
- RNA Velocity - Trajectory inference