RNA Secondary Structure Prediction¤
This example demonstrates how to predict RNA secondary structure using DiffBio's differentiable RNA folding operator.
Overview¤
RNA secondary structure (base pairing patterns) is critical for:
- Understanding RNA function
- Designing antisense oligonucleotides
- Predicting miRNA targets
- Analyzing ribozymes and riboswitches
DiffBio implements the McCaskill partition function algorithm in a differentiable manner, enabling gradient-based optimization of RNA structure predictions.
Prerequisites¤
import jax
import jax.numpy as jnp
from flax import nnx
from diffbio.operators.rna_structure import (
DifferentiableRNAFold,
RNAFoldConfig,
)
Step 1: Encode RNA Sequence¤
RNA sequences use one-hot encoding similar to DNA:
# RNA sequence (should form a stem-loop)
rna_str = "GCGCAAUAGC"
# Encode: A=0, C=1, G=2, U=3
nuc_map = {"A": 0, "C": 1, "G": 2, "U": 3}
rna_indices = jnp.array([nuc_map[n] for n in rna_str])
rna_seq = jax.nn.one_hot(rna_indices, 4)
print(f"RNA sequence: {rna_str}")
print(f"Sequence length: {len(rna_str)}")
print(f"Encoded shape: {rna_seq.shape}")
Output:
Step 2: Create RNA Fold Predictor¤
# Configure the folding predictor
config = RNAFoldConfig(
temperature=1.0, # Boltzmann temperature (1.0 = 37°C)
min_hairpin_loop=3, # Minimum hairpin loop size
)
rngs = nnx.Rngs(42)
predictor = DifferentiableRNAFold(config, rngs=rngs)
Step 3: Predict Structure¤
# Predict secondary structure
data = {"sequence": rna_seq}
result, _, _ = predictor.apply(data, {}, None)
bp_probs = result["bp_probs"]
log_z = result["partition_function"]
print(f"Base pair probability matrix shape: {bp_probs.shape}")
print(f"Log partition function: {float(log_z):.4f}")
Output:
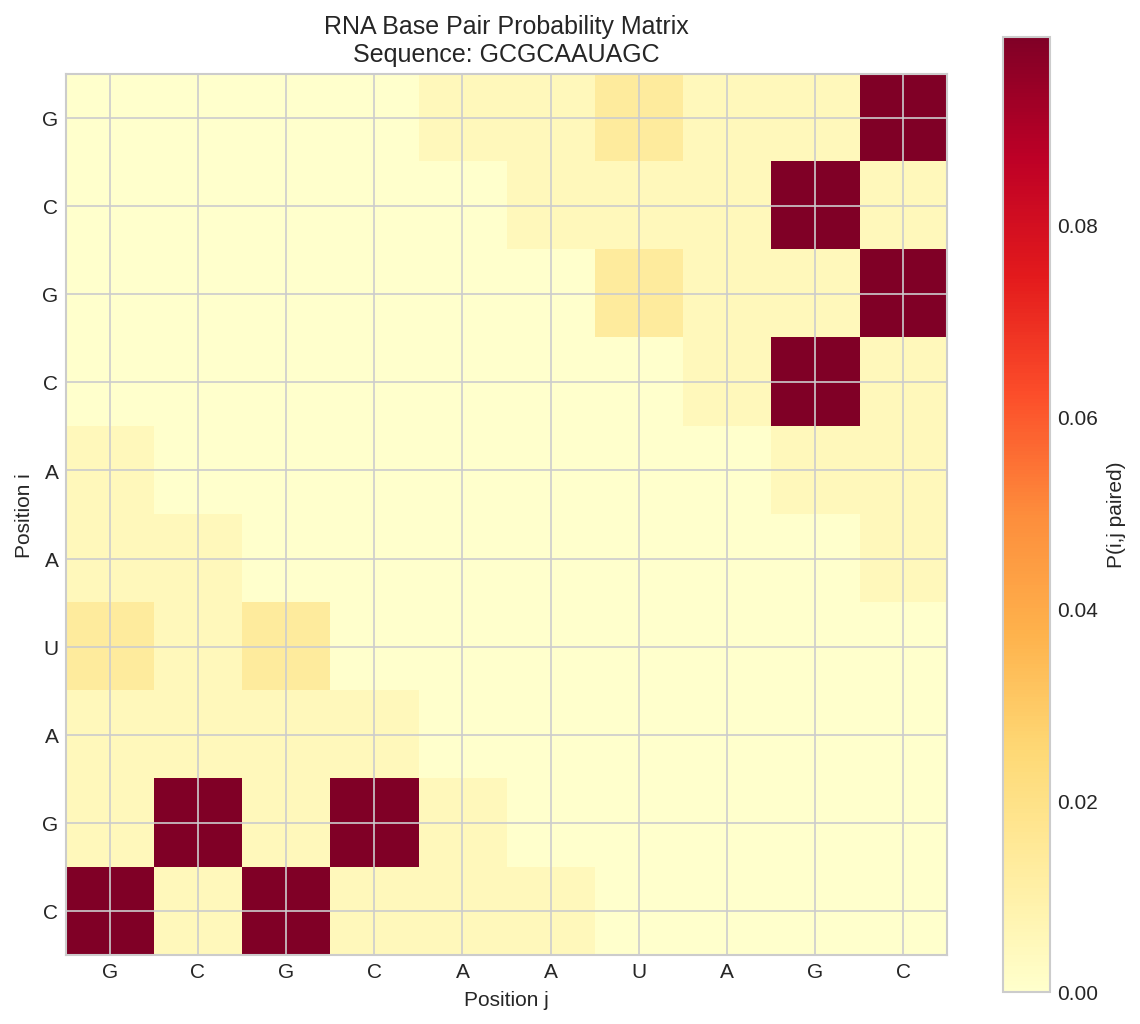
Base pair probability matrix showing the probability of each position pairing with another. Higher values (brighter colors) indicate more probable pairs.
Step 4: Analyze Base Pair Probabilities¤
Find high-probability base pairs:
# Find high-probability base pairs
threshold = 0.01 # 1% probability threshold
print(f"\nHigh probability pairs (p > {threshold}):")
for i in range(len(rna_str)):
for j in range(i + 1, len(rna_str)):
prob = float(bp_probs[i, j])
if prob > threshold:
print(f" {rna_str[i]}{i+1}-{rna_str[j]}{j+1}: {prob:.4f}")
Output:
High probability pairs (p > 0.01):
G1-U7: 0.0135
G1-C10: 0.0997
C2-G9: 0.0997
G3-U7: 0.0135
G3-C10: 0.0997
Understanding the Output¤
Base Pair Probability Matrix¤
The bp_probs matrix contains the probability \(P(i,j)\) that positions \(i\) and \(j\) form a base pair:
where \(Z\) is the partition function and \(Z_{ij}\) is the partition function over structures containing the (i,j) base pair.
Partition Function¤
The log partition function log_z measures the total statistical weight of all possible structures:
A larger partition function indicates more possible structures (more structural flexibility).
Visualizing Structure¤
Convert probabilities to dot-bracket notation:
def probabilities_to_structure(bp_probs, threshold=0.5):
"""Convert base pair probabilities to dot-bracket notation."""
n = bp_probs.shape[0]
structure = ['.'] * n
# Find pairs above threshold
for i in range(n):
for j in range(i + 1, n):
if bp_probs[i, j] > threshold:
structure[i] = '('
structure[j] = ')'
return ''.join(structure)
# Get predicted structure
predicted_structure = probabilities_to_structure(bp_probs, threshold=0.05)
print(f"Sequence: {rna_str}")
print(f"Structure: {predicted_structure}")

Arc diagram visualization of RNA secondary structure. Arcs connect base pairs with arc height proportional to pairing probability.
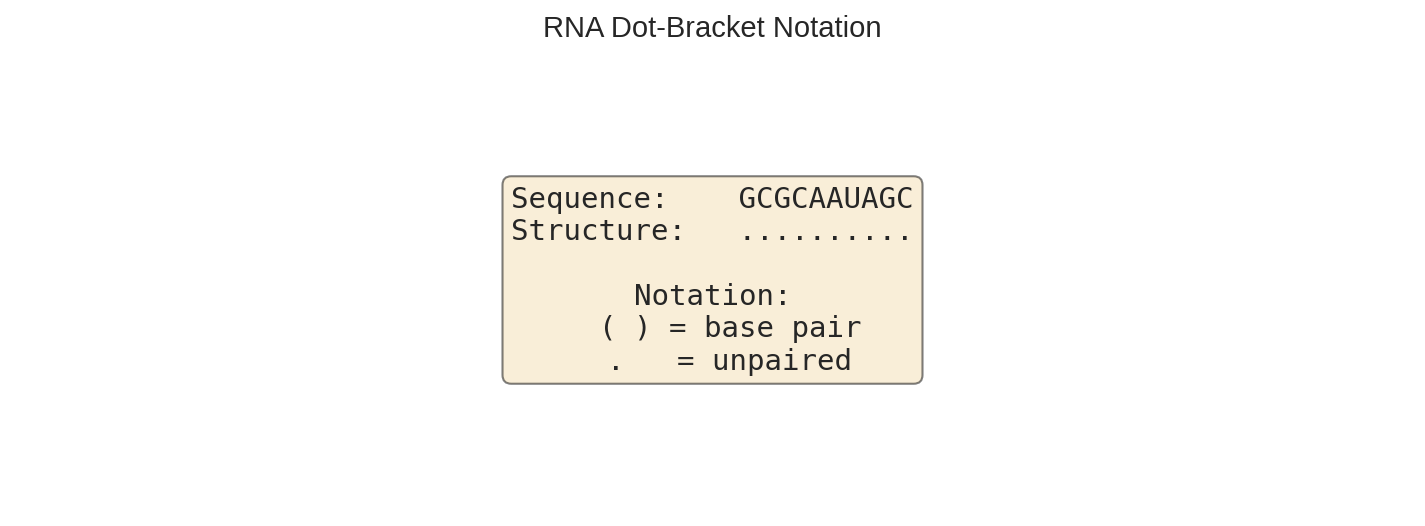
Dot-bracket notation with paired bases shown as parentheses and unpaired bases as dots.
Differentiability¤
The RNA folding operator is fully differentiable:
def structure_loss(predictor, data):
"""Loss function based on structure."""
result, _, _ = predictor.apply(data, {}, None)
# Maximize partition function (minimize folding energy)
return -result["partition_function"]
# Compute gradients
grads = nnx.grad(structure_loss)(predictor, data)
print("Gradient computation: SUCCESS")
This enables:
- Sequence design: Optimize sequences for desired structures
- Thermodynamic parameter learning: Learn folding parameters from data
- End-to-end RNA analysis: Integrate folding into larger pipelines
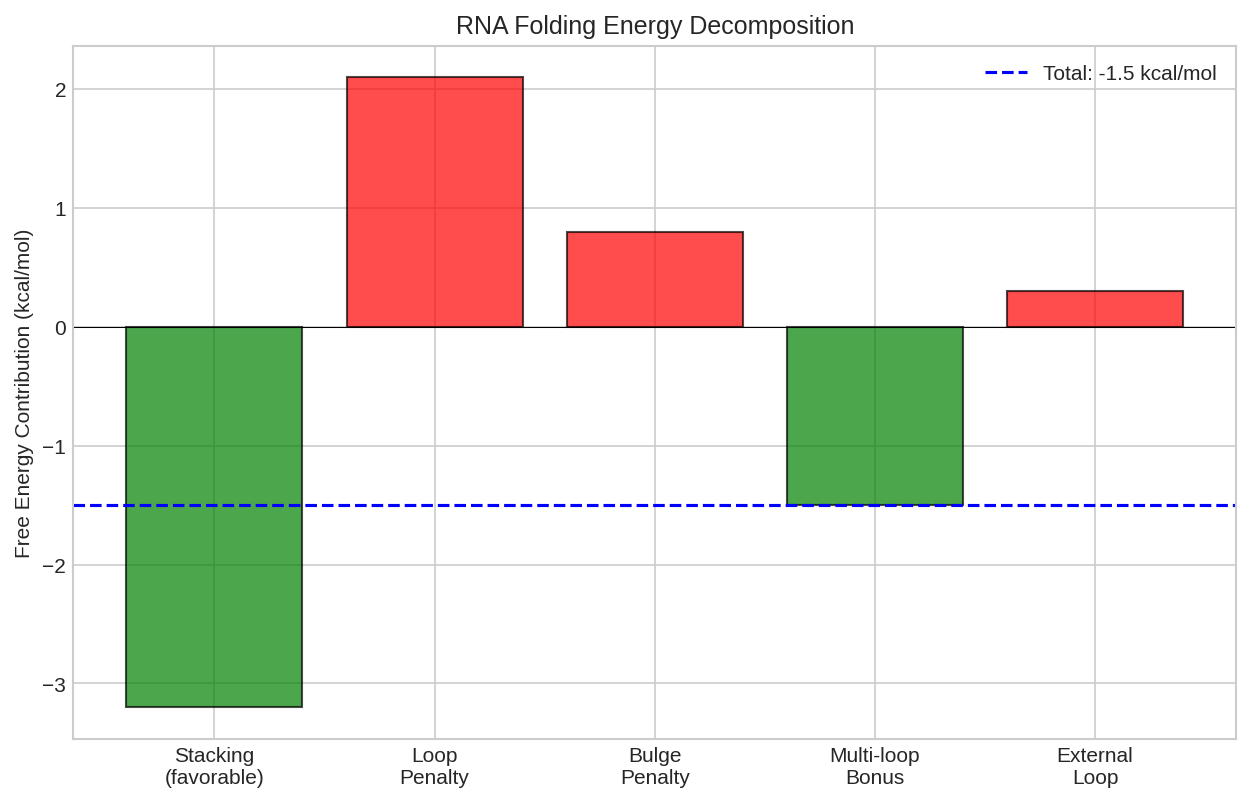
Partition function contributions showing how different structural features contribute to the total statistical weight.
Valid Base Pairs¤
The algorithm considers standard Watson-Crick and wobble pairs:
| Pair | Type |
|---|---|
| A-U | Watson-Crick |
| G-C | Watson-Crick |
| G-U | Wobble |
Configuration Options¤
| Parameter | Description | Default |
|---|---|---|
temperature |
Boltzmann temperature (1.0 = 37°C) | 1.0 |
min_hairpin_loop |
Minimum unpaired bases in hairpin | 3 |
Next Steps¤
- Protein Secondary Structure - Predict protein structure
- HMM Sequence Model - Sequence models with hidden states
- DNA Encoding - Encode sequences for other operators